【トピックス】
ColabReaction:反応の核心に、誰でも飛び込める時代へ
佐藤玄、唐澤昌之、寺田透、レオチーシャン、矢島英明、荒井秀太、西﨑博光
東大院・農、東大院・農、東大院・農、山梨大院・工、山梨大院・工、山梨大院・工、山梨大院・工
1.UMAの衝撃、ColabReactionの誕生
──反応機構解析に訪れた転換点
2025年5月14日、Meta社が発表した大規模分子データセット「Open Molecules 2025 (OMol25)」1)と、それに基づいて構築された機械学習ポテンシャル「Universal Model for Atoms (UMA)」2,3)は、計算化学界に大きな衝撃をもたらした。UMAは、従来の密度汎関数理論 (DFT) 計算と同等の計算精度を維持しながら、桁違いの計算速度を実現しており、今後の計算化学的な反応機構解析の進め方そのものを根本からか塗り替えかねないポテンシャルを秘めている。UMA発表からわずか26日後の2025年6月9日、我々はUMAとダブルエンド型探索手法「Direct MaxFlux (DMF)」4–6)を組み合わせた新たな遷移状態構造探索手法を世界に先駆けて公開した7)。さらに、同手法をGoogle Colaboratory上で誰もが使える形に実装し、 ColabReaction8)として2025年7月14日にオンラインで無償公開した9,10)。これにより、計算化学に不慣れな実験系の研究者や高性能な計算資源を持たない学生でも、反応機構解析を手軽に行える計算環境が整った。分子レベルの反応機構解析を実験の合間に気軽に行うことができる時代が来たとも言うことができる。本稿では、ColabReactionの開発背景、および実際の応用例を交えつつ、本手法の有用性と今後の反応機構解析研究の展望について概説する。

2.経験に頼る時代の終焉
──遷移状態探索、自動化と高速化の現在地
近年、量子化学 (QM) 計算をはじめ、QM/MM法、MDシミュレーション、ab initio MD法など、多くの理論手法が酵素反応機構解明に使われることが一般的になってきた。これにより、実験だけでは見えない一連の反応過程が理論的に可視化されるようになり、より詳細に反応機構を理解することが可能となった。また、トップジャーナルに論文を掲載する上で、実験データに加えて理論計算による裏付けを行うことは「補助的」なデータではなく事実上「必須」となってきており、計算化学の需要は高まってきている。
しかし、遷移状態 (TS) 構造は目に見えないものであり、その探索は非常に困難であり、TS探索には依然として大きな障壁が存在する。DFTレベルでの構造最適化は計算コストが高く、計算の実施にも専門知識と経験が求められる。従来主流であったシングルエンド型手法では、TSに近い構造を人の手で地道に探索し、それを初期構造として構造最適化を行う必要があった。Gaussianのmodredundantオプションなどを駆使して原子間距離や角度を微調整し、最適化結果を何度もチェックする――そうした繰り返し作業が不可欠だった。特に、複数の結合生成や解離が同時に起きるような協奏的反応系では、構造探索の難易度は著しく高く、多くの場合専門家の「職人技」に頼るしかなかったのが実情であった。そのため、反応の核心に迫る一歩手前で、多くの研究者が立ち止まってしまうことがしばしばあった。
こうした問題に対処すべく、深い専門知識を持たなくても TS探索を行うことができる手法であるダブルエンド型のTS探索手法が有力な選択肢として長年研究されてきた。ダブルエンド型探索法は、反応物 (reactant) と生成物 (product) の構造を入力とすることで、プログラムが自動的にTS構造を導出する手法である。計算理論の詳細は本稿では割愛するが、この方法により専門家の「職人技」に頼る必要はなくなり、TS探索の自動化を行うことができる。しかし、エネルギーや力 (Force) の計算は外部の量子化学計算ソフトに依存するため、用いる理論レベルによっては結局のところ速度的なボトルネックが残るため、「自動化できても遅い」というジレンマを抱えていた。
この壁を乗り越える鍵となったのが、機械学習 (ML) ポテンシャルの導入である。MLポテンシャルとは、事前に大量の分子についてDFT計算を行い、その構造とエネルギー・力の関係性を学習させたモデルである。任意の分子構造を入力すると、即座にそのエネルギーや力を推定できる。原理的には、DFTと同等の精度を維持しながら、計算時間は数桁レベルで短縮可能である。これまでにDeepMDやANIをはじめ、数多くのMLポテンシャルが提案されてきた。日本発の代表的な例としては、Preferred Networks社が販売した Matlantisが挙げられる。DeepMDやMatlantisは、材料科学や無機化学分野への応用を念頭に設計されており、主にエネルギーおよび力 (Force) の高精度な予測を可能にする原子スケールポテンシャルとして知られている。一方、有機分子や生体分子に特化した汎用的な学習データが限られていたため、柔軟な構造変化や複雑な反応性の再現においては課題が残るケースも見られた。また、ANI-1x (有機分子向け)、CHGNet (格子欠陥や電荷密度の予測)、DPA-2 (反応経路の予測支援)、MACE (対称性を保った原子環境表現)、Orb-v2 (軌道ベースのポテンシャル構築) など、多様な MLポテンシャルや関連手法が開発されてきた。しかし、これらの多くのモデルにおいては、学習データの網羅性、遷移状態や反応中間体の精度、化学空間の偏り、あるいは実運用の難易度など、適用範囲を広げる上で解決すべき課題が依然として存在している。
そこへ登場したのが「OMol25」である。Meta社が2025年に公開したこのデータセットは、分子科学分野で前例のない規模と網羅性を誇る。具体的には以下のよう
な特徴を持つ:
・ 約1億件の高精度な分子構造データ (計算レベル:ωB97M-V/def2-TZVPD)
・周期表の大部分にあたる83種類の元素をカバー
・最大350原子規模の大分子まで含む、多様な構造群
単なる「量」のインパクトにとどまらず、含まれる化合物の種類や構造の多様性においても、従来の限界を大きく超えている。Meta社はこれまでにも、OC20 (触媒反応、2020年)、ODAC23 (CO2吸着、2023年)、OMat24 (無機材料、2024年) といった大型データセットを次々と公開してきた。UMA (Universal Model for Atoms) は、これらのデータ群を統合的に学習することで構築された、かつてない汎用性を持つ MLポテンシャルモデルである。
このUMAと、最新のダブルエンド型TS探索手法DMFを組み合わせたのがColabReactionである。ColabReactionは、Google Colaboratory上で動作する設計となっており、GPUマシンもローカル環境も必要ない。必要なのは、ネット環境と少しの好奇心だけだ!これまで反応経路の理論解析をあきらめていた研究者たちにとって、ColabReactionは「見えないもの (遷移状態) を、見つける」ための突破口となる。
3. 多様な反応への展開:ColabReaction はどこまで使えるのか?
本稿では、ColabReactionの実際の適用例として、以下の3種類の酵素反応モデルを対象に検証を行った:①P450水酸化反応、②テルペン環化酵素における協奏的反応、③Diels–Alder反応。これらはそれぞれ、ラジカル反応、極性反応、ペリ環状反応という異なる反応機構に分類され、ColabReactionの汎用性と精度を評価する上で適切なテストケースであると考えた。なお、今回はモデル反応のため基質特異性については考慮していない。
各反応について、反応物および生成物の構造を入力し、ColabReactionにより遷移状態構造の探索を実行した。計算はすべてGoogle Colaboratoryの無料プランで提供されるNVIDIA T4 GPU環境上で行い、いずれのケースにおいても10分以内で計算を完了することができた。これは、従来のDFT計算に基づく手法では1件あたり数時間〜1日以上を要していたのに対し、100倍以上の計算速度向上に相当する。
性能指標として、活性化エネルギーに関する平均絶対誤差 (MAE) は2.6 kcal mol-1、遷移状態構造の RMSDはわずか 0.40 Åであり、実用に耐える精度を十分に満たしていることが確認された。これらの結果を図1に示す。
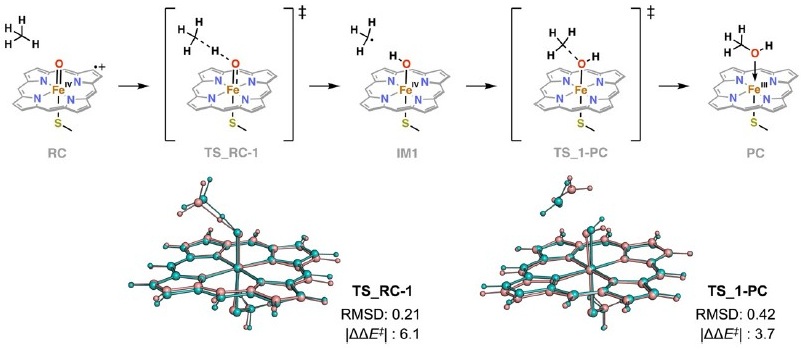
図1 ColabReactionを用いた酵素反応の計算例①:P450水酸化反応
次に、ColabReactionの応用範囲をさらに拡張すべく、より複雑な酵素反応機構の解析に取り組んだ。具体的には、trichobrasilenol生合成に含まれる協奏的転位反応を対象とし、連続する結合再編成が一つの遷移状態構造で記述されるような反応経路を再現可能か検証した (図2)。この反応は、従来のシングルエンド型探索手法では初期構造の探索が難しく、探索に熟練した専門知識が求められることで知られている。ColabReactionを用いた探索では、特別なパラメータ調整なしに高精度なTS構造が得られ、複雑な酵素反応にも応用可能であることが示された。
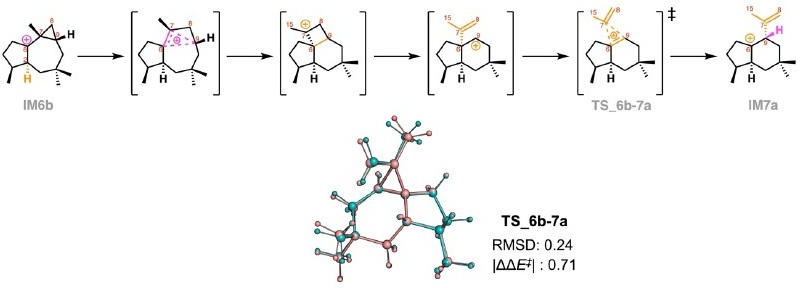
図2 ColabReactionを用いた酵素反応の計算例②:テルペン環化酵素における協奏的反応
さらに、bisorbicillinoid生合成に含まれる Diels–Alder反応についても解析を行った (図3)。この反応は一見協奏的に見えるが、実際には二つの結合生成が段階的に進行することが多く、「energetically concerted, bonding stepwise」として分類される。従来の遷移状態探索では、どの結合生成に沿って座標を定義すべきかが明確でない場合、構造探索が困難になることが多かった。ColabReaction では、反応物および生成物の構造のみを入力すれば、結合順序に依存せず、自動的かつ高速に最適な遷移状態構造が導出される。図3に示すとおり、得られた構造はDFT計算と比較して遜色なく、しかも計算時間は約10分と極めて短時間であった。
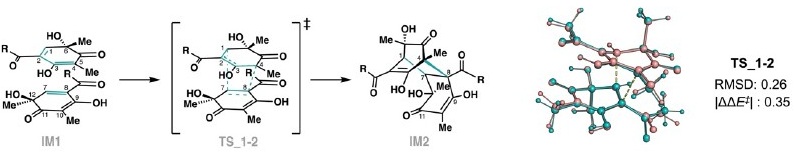
図3 ColabReactionを用いた酵素反応の計算例③:Diels–Alder反応
以上の結果から、ColabReactionは単なる速度向上だけでなく、反応機構の多様性に対する柔軟な対応力と、専門知識に依存しない高い汎用性を兼ね備えていることが明らかとなった。これは、これまで困難とされていた複雑な有機反応や酵素反応に対しても、計算化学的アプローチを日常的に導入できる道を拓くものである。
4.はじめてのColabReaction:構造入力からTS探索まで
ColabReactionは、専用ウェブサイト (https://ColabReaction.net) から誰でも無償で利用可能である。ソースコードはGitHub上で公開されており10)、詳細なユーザーガイド11)も整備されている。
I. Setup Section
まず、図4に示すように、反応物 (reactant) および生成物 (product) の三次元構造ファイルをアップロードする。対応ファイル形式は .xyz、.com、.gjf、.pdb、.mol、.sdfであり、一般的な分子構造形式に広く対応している。

図4 反応物と生成物ファイルのアップロード画面
ただし、ダブルエンド型の探索手法においては、反応物と生成物間で対応する原子の順序 (index) が一致している必要がある点に注意が必要である。これは、反応座標の補間において原子間の対応付けが前提となるためである。
ColabReactionでは、PythonのPanelライブラリを用いたユーザーインターフェース (UI) を実装しており、ファイルはドラッグ&ドロップ操作で簡便にアップロード可能である。アップロード後は、各構造の三次元表示および原子のindexが即座に可視化され、ユーザーが原子の順番をその場で確認できる設計となっている。
次に、電荷 (charge)、スピン多重度 (spin multiplicity)、イメージ数 (nmove, デフォルト値20)、評価点の自動更新 (update_teval, デフォルト値OFF)、および収束条件 (convergence, デフォルト値tight) を設定する。入力が完了したら、「Apply」ボタンをクリックする。さらに、UMAを使用するためには、Meta社が提供する Hugging Faceアクセストークンの入力が必要となる。事前に Hugging Face上のUMAページからアカウント登録およびアクセストークンの取得を行い、ColabReactionに入力する必要がある。
II. Execution Section
すべての設定が完了したら、実行セクション (Execution Section) に進む。「ランタイム」メニューから「現在のセルとその下のセルを実行する」を選択することで、反応経路の探索、エネルギー計算、構造最適化、振動解析、エネルギーダイヤグラムの作成 (図5)、出力ファイルのダウンロードまでの一連の処理が自動的に開始される。
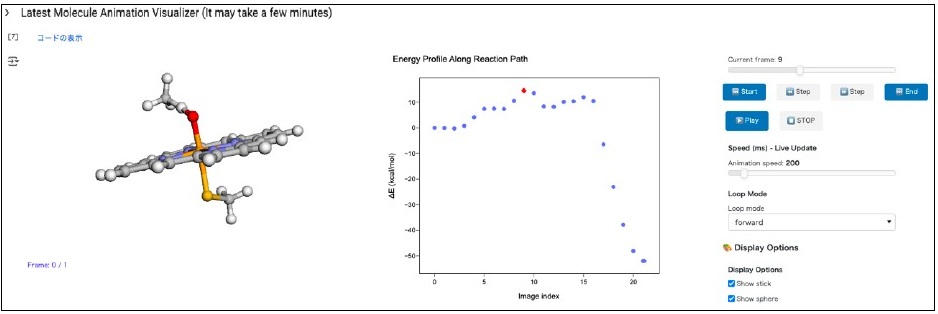
図5 計算結果の表示画面
ColabReactionでは、得られた反応経路に沿ってエネルギーダイアグラムを生成し、各極大点において数値的ヘシアンに基づく振動解析を行う。これは、UMAから得られた力 (Force) 情報を用いてヘシアン行列を近似的に導出する手法であり、DFT計算に基づく解析的なヘシアンに比べて精度は限定的ではあるものの、概ね整合的な振動モードが得られることが多い。振動モードをアニメーションとして視覚的に確認することで、目的の遷移状態が物理的に妥当な構造であるかを迅速に判断できる。望みの振動モードを持つ各極大点を初期構造として DFT計算によるTS最適化とIRC計算に進むことができる。ColabReactionは、機械学習ポテンシャルによる高速・自動化計算と、従来法との橋渡しを担う補助的ツールとして位置付けられる。
5. 遷移状態を、すべての研究者の手の中に
ColabReactionの登場は、まさに「反応機構解析の夜明け」を告げるものである。従来、遷移状態構造の探索は専門家の手によって一件ずつ慎重に、そして多くの時間と計算資源を投じて行われてきた。その結果、計算化学はごく一部の専門家に限られた「特権的な道具」としての側面を持ち続けていた。しかし今、DMF/UMA法1-7)を中核に据えたColabReactionの登場により、その状況は根底から覆されようとしている。Google Colaboratoryという誰もがアクセス可能なプラットフォーム上で、専門的なセットアップや高価なソフトウェアの購入なしに、即座に高精度な反応経路解析を始めることができる。これは単なる技術的進歩ではない。研究者の誰もが、実験の補完として反応機構解析を“日常的に”活用できる新時代が幕を開けたということである。こうした「TS構造探索の民主化」は、かつて構造生物学の分野で起こった革新とも共鳴する。たとえば、AlphaFoldの登場は、タンパク質立体構造予測における長年のボトルネックを一挙に解消し、構造生物学に革命をもたらした。さらに、ColabFoldのようなクラウドベースの実装により、誰でも気軽にブラウザ上から数分で高精度な構造予測を実行できるようになった。AlphaFold3では複合体予測やリガンド結合予測にも対応が進んでおり、もはや“構造予測は一部の専門家だけの仕事”ではなくなった。このような民主化が、創薬、機能解析、酵素設計など多様な分野に革新的な進展をもたらしているのは、言うまでもない。同様の例は材料科学にも見られる。マテリアルズ・インフォマティクス (MI) は、膨大な材料候補を一つ一つDFTで計算していては現実的でなかった探索空間に対し、機械学習を導入することで、材料開発の速度と効率を劇的に高めてきた。特に電池材料や触媒設計では、これまで数年単位で進められていた開発が、わずか数ヶ月、あるいは数週間で達成される例も珍しくなくなってきている。
ColabReactionがもたらす変化は、既存の技術革新と同様に、研究の在り方そのものを塗り替える可能性を秘めている。たとえば、DFT計算による反応機構解析が特に盛んに行われてきたテルペン環化酵素でさえ、これまでに報告された計算化学の論文数はおよそ 100報程度にとどまる。これは、TS構造探索・最適化、IRC計算、エネルギーダイヤグラムの描画といった各工程に、多大な計算コストと労力を要していたことが大きな要因である。ColabReactionによって、これら一連の作業が飛躍的に高速化されたことで、これまで困難とされていた網羅的な反応経路の探索や、反応機構に関する大規模なデータセットの構築も現実味を帯びてきた。実験科学と計算化学という違いはあるが、こうしたデータの集積は、かつてトランスクリプトームやメタボローム解析が引き起こしたオミクス分野の爆発的発展を想起させる。遷移状態構造を体系的に蓄積・解析することで、個別事例の積み上げでは捉えきれなかった反応機構の共通パターンや設計原理が浮かび上がる可能性もある。とりわけ酵素工学の分野においては、これまで経験則に依存してきた機能改変戦略に対し、構造と反応性に基づく新たな論理的アプローチが導入される契機となるだろう。
ColabReactionは、単なるツールではない。これは、 あらゆる研究者が“反応の中を見通せる”という選択肢を等しく持てるようになる、新しい時代の旗印である。計算化学は、専門家の「職人技」から、開かれた研究基盤へと変貌しつつある。その変化を牽引する第一歩が、今ここにある。「反応を可視化する」ことが、「反応の内側に飛び込む」ことが、そして「反応を自らの手で設計する」ことが、いよいよ誰にとっても当たり前の研究手法になっていく――その入口として、ColabReactionが多くの研究者の手に届くことを願っている。
謝辞
本研究は、JSPS科研費 (課題番号24K01626、22H05125 (佐藤玄)、22H05126 (寺田透)、25H01577 (レオチーシャン))、日本医療研究開発機構 (AMED) 生命科学・創薬研究支援基盤事業 (BINDS) (課題番号 JP25ama121027 (寺田透)) の支援を受けたものです。
文献
1) Levine, D. S., Shuaibi, M., Spotte-Smith, E. W. C., Taylor, M. G., Hasyim, M. R., Michel, K., Batatia, I., Csányi, G., Dzamba, M., Eastman, P., Frey, N. C., Fu, X., Gharakhanyan, V., Krishnapriyan, A. S., Rackers, J. A., Raja, S., Rizvi, A., Rosen, A. S., Ulissi, Z., Vargas, S., Zitnick, C. L., Blau, S. M., Wood, B. M.: The Open Molecules 2025 (OMol25) dataset, evaluations, and models, (2025).
2) “UMA: A Family of Universal Models for Atoms.”: <https://ai.meta.com/research/publications/uma-afamily-of-universal-models-for-atoms/>, cited 21 June, 2025.
3) “fairchem: FAIR Chemistry’s library of machine learning methods for chemistry,” Github: https://github.com/facebookresearch/fairchem.
4) Koda, S.-I., Saito, S.: J. Chem. Theory Comput., 20, 2798 (2024).
5) Koda, S.-I., Saito, S.: J. Chem. Theory Comput., 21, 3513 (2025).
6) Koda, S.-I., Saito, S.: J. Chem. Theory Comput., 20, 7176 (2024).
7) Nakano, M., Karasawa, M., Ohmura, T., Terada, T., Sato, H.: ChemRxiv (2025). DOI: 10.26434/chemrxiv-2025-md8k6-v2.
8) Karasawa, M., Leow, C. S., Yajima, H., Arai, S., Nishizaki, H., Terada, T., Sato, H.: ChemRxiv (2025). DOI: 10.26434/chemrxiv-2025-zvkqk
9) https://ColabReaction.net
10) https://github.com/BILAB/ColabReaction
11) https://bilab.github.io/ColabReaction/User_Guide_JP.pdf
![]()