【トピックス】
人工非ヘム金属酵素における柔軟性の高い配位挙動の発見
三枝直樹、藤枝伸宇
阪公大院・農
1.はじめに
化石資源の節約や環境汚染抑制の観点から、効率的かつ環境に配慮した物質変換方法が求められている。そのため、無機、有機、超分子など幅広い分野の研究者がそれぞれ得意とする合成分子を基盤とした触媒を設計し、様々な反応系の向上を行ってきた。その中でも近年、注目を集めている触媒として人工金属酵素 (ArM) が挙げられる1)。ArMはタンパク質に、相互作用可能な金属錯体や遷移金属イオンを取り込ませることで構築するバイオ触媒である。タンパク質骨格をキラルな反応場として用いることで、反応の立体制御が可能になると期待されている。以下に説明するようにArMの設計には多数の方法が存在する。1つはヘムの置換を用いたArMである2,3)。多くの天然金属酵素は、ヘムのような金属含有補因子を活性部位に結合している。そこで天然の金属補因子を合成誘導体に置換することで、金属酵素が触媒できる反応の範囲拡張が達成されてきた。ミオグロビンを骨格とし、天然のヘムをイリジウムといった貴金属を含む様々なポルフィリン錯体に置換することで、カルベン挿入とシクロプロパン化の両方を優れたエナンチオ選択性で触媒することが示された3) (図1A)。また、超分子相互作用を利用したArMも報告されている。代表的なものとして、ストレプトアビジンとビオチンの非常に強い相互作用を用いたArMが挙げられる4,5)。実際に、ビオチン化したイリジウム錯体を用いたArMは、炭素–水素結合へのカルベン挿入反応や、光還元反応など、非天然の反応を高い立体選択性で実現した4)。また、4Fe–4Sクラスターの埋め込みにより、人工的なFischer–Tropsch触媒として二酸化炭素の還元に成功した例も報告され5)、その汎用性と応用性の高さが注目されている。ここまでは合成された金属錯体を用いるArMを説明してきたが、直接金属イオンを配位させることでも構築できる。タンパク質のヒスチジン、システイン、アスパラギン酸、グルタミン酸などの配位性アミノ酸を利用するため、遊離の金属イオンを直接用いることができ、非常に簡便である。例えば、細菌の外膜タンパク質であるOmpFを用いたArMが報告されている6)。以上のようにArMは、天然酵素の持つ高い選択性と穏やかな反応条件という利点をベースに非天然反応を可能にする強力なツールである。
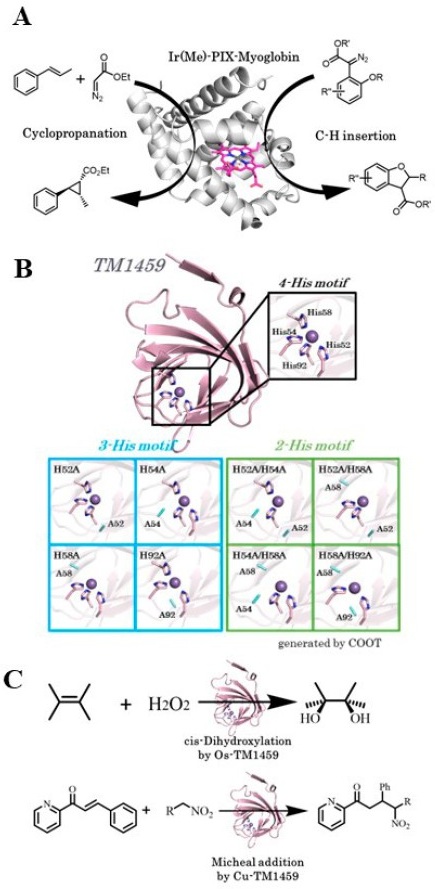

我々のグループではThermotoga maritima由来のホモ二量体タンパク質TM1459を用いてArMを構築した (図1B)。このタンパク質はcupinスーパーファミリーに属し、分子量が12,977 Daと小さく、好熱菌由来であることから安定で扱いやすくArMの土台分子として適している。また、4つのヒスチジンからなる金属結合部位を有しており、金属を入れ替えることで種々の活性を付与することができ、ある意味では高分子金属配位子として使用可能であるとも言える。オスミウムを結合させたTM1459は位置選択的なcis-1,2-ジヒドロキシ化反応を触媒した7) (図1C)。さらに金属結合部位であるヒスチジン残基に変異を導入することで3つのヒスチジンからなる3組残基構造 (3-Hisトライアド) や2つのヒスチジンからなる2組残基構造 (2-Hisダイアド) といった異なる配位構造の金属結合モチーフに変換することができる (図1B)。つまり、様々な配位構造を有したライブラリーとして変異体をスクリーニングすることによって、より良い人工金属酵素を探索することができる。このようにして得られた銅結合型TM1459変異体は、α, β-不飽和ケトンのモデル基質として、2-アザカルコンを用いたニトロアルカンの立体選択的マイケル付加反応を触媒した8) (図1C)。これらの反応に用いたTM1459の結晶構造を取得した所、金属結合部位が予想通りの3配位や、2配位の構造であることが示された。またドッキングシミュレーションにより、基質結合様式と高い選択性の相関が説明できた。
2.天然金属酵素
金属酵素は活性中心に金属補因子を持つ。特に、アミノ酸残基に直接金属イオンが結合した活性中心をもつものを非ヘム金属酵素と呼ぶ。この酵素の金属補因子は、触媒サイクルにおいて位置や配位構造があまり変化しないというのが通説であった。しかし近年の分析技術の発展により、一部の非ヘム金属酵素では金属補因子の位置遷移が見られ、機能的役割を持つ例が報告されている9-11)。D-キシロースイソメラーゼは2つの二価金属イオンを触媒作用に利用し、M-1部位は固定であるのに対して、もう一方のM-2部位は1.8 Åの位置遷移が観測された10)。M-2部位はこの遷移により、基質である開環型のキシロースの安定化と異性化反応の中心的な段階である、ヒドリドシフトを誘発するために不可欠な役割を持つと言われている。他にもクラスⅡピルビン酸依存性アルドラーゼにおいて、活性状態と静止状態の間で2.3 Åの金属中心が位置遷移することが知られている11)。活性中心では基質となるヒドロキシピルビン酸が結合することで金属は活性状態へと変化する。したがって、145、D171、S116′で配位した、奥側に埋もれた休止状態から、E145、D171および基質で配位する、活性部位の浅い位置に移動した活性状態へと変化することが示された (図2)。このように天然酵素において、まだ観測例は少ないものの、金属中心の位置遷移は触媒作用や活性制御に寄与していることが明らかとなりつつある。
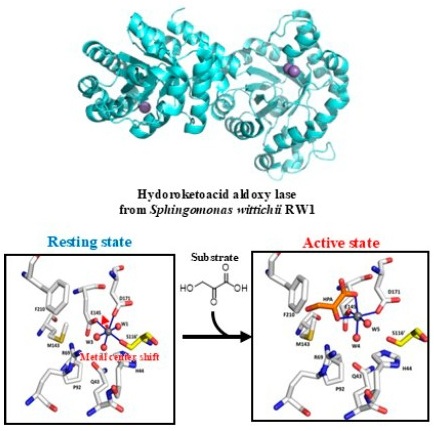
![]()
我々が行ってきたTM1459を用いたArM設計において、触媒活性を向上させる基本的な戦略は金属の種類や第一配位圏のスクリーニングである。我々はこの設計戦略に従い、異なる反応や化学修飾による新たな金属配位場を創出した。その結果、良好な反応性を示したArMにおいて、ここで述べたような金属中心の位置遷移が観測された。一般的には金属酵素において触媒金属は結合位置を強く固定されていると考えられてきた。しかし、上で述べたように、天然酵素において金属の位置遷移により反応性を向上させる例が報告され始めた。この金属の動的挙動という現象の制御はAIなどによる予測がまだ行えない領域であり、次の世代のArM設計においても新たな選択肢になる可能性がある。そこで、本稿では金属の位置遷移が見られた2つの例を示したい。
3-1 HHEトライアドを金属中心とする人工金属酵素12)
1つ目は金属結合モチーフとして、2つのヒスチジンとカルボン酸をもつアミノ酸、つまりグルタミン酸またはアスパラギン酸 (His-His-Glu[HHE]またはHis-His-Asp[HHD]) を導入したTM1459変異体である。特に、金属中心の3つの配位座をアミノ酸が占有するとき、同じ平面上になるmeridional (mer)と同じ方向に集中するfacial (fac)の2種類が現れる。天然では後者しか見られていないことから、我々が着手する前から、すでにHHD fac-トライアドを持つArMはいくつか報告されていた。例えば、Reetzらは元々金属タンパク質でないTIMバレルタンパク質を骨格に持つイミダゾールグリセロールリン酸シンターゼ内にHHD fac-トライアドを導入し、銅を配位させることで人工Diels-Alderaseを構築した13)。これは2-アザカルコンとシクロペンタジエンの反応を触媒し、73%の変換率と良好なエナンチオ選択性 (エナンチオマー過剰率ee=46%、ジアステレオ選択性 (エンド:エキソ=93:7) ) を示した。一方、Songらは、タンパク質表面の変異により単量体タンパク質を4量体化させることで、人工的にβ−ラクタマーゼを構築した。この多量体化によって生じた新たなキャビティに亜鉛を結合するHHE構造が構築された14)。このような先行研究に触発され、HHEを持ったTM1459を調製することで2-アザカルコンと、ジメチルマロン酸やアセト酢酸メチルといった活性メチレン化合物とのマイケル付加反応を触媒させるArMの構築を目指した (図3A)。そこで金属に配位するヒスチジンに対して、HHEを構成できるように変異を加え、さらなる金属結合部位の多様化を実現した。その中でH52A/H58E変異体はアセト酢酸メチルでの反応においてee=96%を達成した。
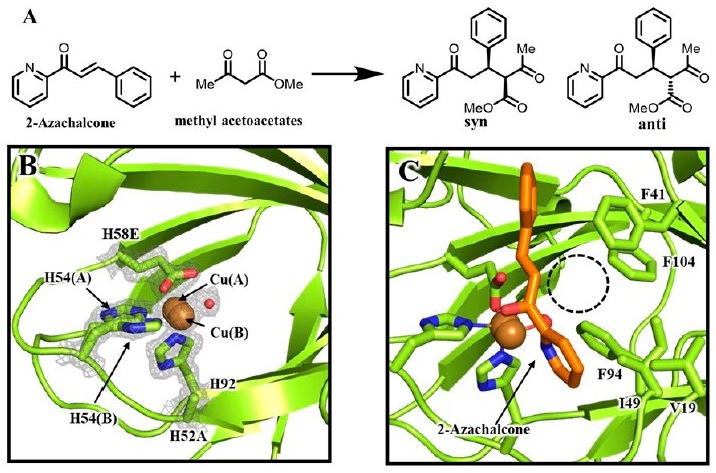

エナンチオ選択性の向上を説明するため、Cu-H52A/H58E変異体の結晶構造解析を、1.15 Åの分解能で決定した (図3B)。金属結合部位は、設計通りのHHE fac-トライアドを形成していることが確認された。His54とHis92の2つの窒素原子、2つの水分子、そしてGlu58の1つの酸素原子からなるN2O3ドナーセットによる四角錐型配位構造である。ここで3-Hisトライアドを持つH52Aの結晶構造に注目し、H52A/H58Eと比較する。H52Aの銅イオンは2つの水分子と共に3つのヒスチジン残基が配位している。二量体の内、ChainAではHis54、His58、及び2つの水分子がエクアトリアル位に、His92が軸位に位置している。一方で、ChainBではHis58、His92及び2つの水分子がエクアトリアル位に、His54が軸位で結合する。つまり、この配位構造は1つの変異体中に異なる配位構造が存在するために、銅中心と基質である2-アザカルコンとの間に少なくとも3種類の異なる2座配位様式が存在することを裏付けている。これが立体選択性の低下を引き起こす要因の1つとして考えられる。
一方で高いエナンチオ選択性を示したH52A/H58E変異体では、Glu58がカルボン酸酸素原子を介し単座配位で銅イオンと結合し、残りの酸素原子が銅に配位する水 分子と水素結合を形成していた (図3B)。この構造から判断すると、基質の2-アザカルコンがこの水分子とリガンド交換するのを防ぎ、Glu58のトランス位 (遠位側) からのみ銅にアクセスできるようになる。これにより2-アザカルコンの結合様式が固定化され、キラル平面が決定されると考えられる。
またこの銅中心において、メジャーな銅種 [Cu(A)]に加えてマイナーな銅種[Cu(B)]の電子密度が観察された (図3B)。これは、銅中心が複数の配位状態を持ち、空間的にわずかに位置遷移しうる「柔軟性」を持つことを示唆している。マイナーなCu(B)では、Glu58と銅の配位結合がCu(A)よりもわずかに伸長しており、Cu(A)が2.20±0.03 Åに対しCu(B)では2.73±0.04 Åに変化している。この構造変化によって、立体障害が少ない銅中心Cu(B)が形成されると考えられた。つまり、2-アザカルコンのケトン酸素、ピリジン窒素がそれぞれHis92及びGlu58の反対側に位置する水分子の位置から銅中心、特にCu(B)に配位しやすくなる。これにより立体障害の少ない銅中心が生成されると考えられ、基質のキラル面を決定する特異的な結合様式を固定することで、高いエナンチオ選択性に繋がったと考えられる。
さらに基質の結合様式を、ドッキングシミュレーションを行うソフトウェアであるAutoDock Vinaを用いて予測した (図3C)。最もエネルギーの低い配座において、2-アザカルコンのケトンおよびピリジン部分は、先の予測通り、銅中心にGlu58、His92のトランス位に結合した構造が得られた。点線の円で示す部分に注目すると、この空間はアザカルコンのSi面側にあり、周囲の5つの疎水性アミノ酸残基、Val19、Phe41、Ile49、Phe94、Phe104によって密に充填されている。生成物の立体構造から推測するとアセト酢酸メチルはこちらの面から反応していることが示唆される。この仮説を裏付けるために、これらのアミノ酸残基をより立体的に嵩高いアミノ酸残基に置換することで、アセト酢酸メチルとの相互作用の変化を観測することとした。その結果、F41WおよびF104Wの変異を追加したH52A/H58Eではジアステレオ選択性が発現し、それぞれ62:38、71:29を示し、エナンチオ選択性も維持した。これらはドッキング構造から予測されたSi面からの攻撃を支持する結果であると言える。
H52A/H58E/F104W変異体の基質結合様式を解明するために、結晶構造解析を行い、1.18 Åの分解能で構造を決定した (図4A, B)。銅中心の配位構造はH52A/H58Eと同様であった。Cu(A)-Cu(B)間の距離は、H52A/H58Eと比べて0.3 Å程度伸長した。導入したTrp104とGlu58を架橋するように、2残基間を架橋する位置に新たな水分子が存在していた。つまりF104Wの変異はこの水分子を結合することで、水分子とアセト酢酸メチルのアセチル基ケトンと水素結合を介して相互作用を形成する。その結果アセチル基側からの侵入を促進し、立体障害の大きいTrp104によりメトキシカルボニル側が空洞に侵入することを阻止した可能性を示唆している。
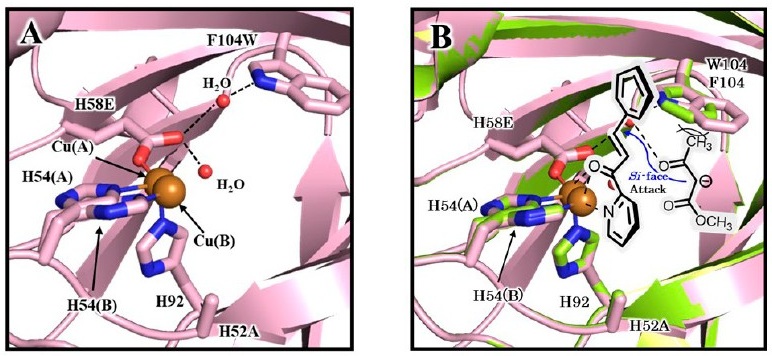

このような特殊な挙動をする銅中心について更なる調査を行うため、H52A/H58Q変異体との比較を行った。H52A/H58Q変異体の結晶構造を決定したところ、Gln58-Cu間の配位結合距離が長く (2.49±0.11 Å)、Cu(A)からCu(B)への距離もH52A/H58Eよりも著しく離れていた (H52A/H58E:0.54±0.02 Å、H52A/H58Q:1.23±0.02 Å)。これにより銅イオンが活性中心で空間的により大きく遷移すること、および銅イオンへの結合親和性が低いこと (解離定数Kdはそれぞれ0.11μM[H52A/H58E]、0.53μM[H52A/H58Q]であった。) が示された。つまり、触媒サイクル中での銅イオンの脱離により、反応性が低下し、収率が著しく減少したと考えられる。以上からH52A、H52A/H58E、H52A/H58Qを比較すると、Cu活性中心の柔軟性は58番目のアミノ酸残基のpKaを反映し、結合の強さに依存した。その中でH52A/H58Eの持つ銅中心がこの反応においてちょうどよい柔軟性であることが示唆された。
以上の結果は、HHE fac-配位銅中心を含有したArMにおいて、天然酵素には見られない反応を触媒する例となった。さらにメジャーな銅種とマイナーな銅種が共存する状態がCu-TM1459変異体ArMにおいても観測され、これが触媒活性の向上につながった可能性が示唆された。このArMのメカニズムに関する詳細な研究は、将来、天然酵素の触媒活性に関してより深い理解につながると考えられる。
3-2 非天然分子を金属結合部位に持つ人工金属酵素15)
2つ目の例は化学修飾による非天然の配位子を導入したTM1459タンパク質である。人工金属酵素の設計において、金属配位能を有する非天然アミノ酸の導入は、天然アミノ酸では不可能な反応を可能にする。Jarvisらは、アンバーコドン抑制技術を用いて、金属配位性非天然アミノ酸の1つである (2,2′-ビピリジン-5-イル) アラニン (Bpy-Ala) を含むArMを合成した16)。その結果得られたCu-ArMは、フリーデル・クラフツアルキル化反応において、良好なエナンチオ選択性を発揮されることを示した。
また選択性を反転させるための戦略として、キラルな小分子触媒のエナンチオマーを利用することやD-アミノ酸からなる小さなメタロペプチドを用いた触媒反応などがある。しかし、D-アミノ酸からなる酵素などの鏡像タンパク質の調製は依然として難度が高い17)。ArMにおいてタンパク質骨格の設計は非常に重要であるが、エナンチオ選択性を制御し、反転させるための活性中心の合理的な設計戦略はいまだ確立されていない。
上で述べたように、我々は以前TM1459タンパク質を用いた2-アザカルコンとニトロアルカンの立体選択的マイケル付加反応について報告した8)。この反応において、エナンチオ選択性の反転を誘起するため、銅結合部位近傍にピリジン配位子を導入する化学修飾について注目した。我々の先行研究ではニトロメタンでの反応において、H52A変異体でS体をee=99%と非常に高い選択性で変換してきた。このH52A変異体の結晶構造では、His54、His58、His92からなる3-Hisトライアドが銅中心に配位し、疎水性ポケットがHis54のトランス位に位置することを示した。この銅中心において、ニトロメタンが、2-アザカルコンのSi面側から結合することを示した。この知見に基づき、H52Aの配位構造の鏡像体を持つ金属結合部位を構築することで、エナンチオ選択性を反転できるという仮説を立てた。
当初、His54のトランス位に位置する疎水性ポケットにヒスチジン残基を導入するアプローチも検討した。しかし、候補としたIle49とPhe94は、銅中心からそれぞれ3.8 Åと6.4 Åの距離にあり、銅に直接配位しない可能性が示唆された。そこで、新しい配位構造を構築するため、4,4′-ジチオピリジン (4-PDS) を用いた化学修飾を行った (図5)。4-PDSは、還元型システインと選択的に反応してジスルフィド結合を形成し、S-(ピリジン-4-イルチオ) システイン (Cys-4py) を生成する。Cys-4pyにおける配位性窒素原子とCα原子の間の距離 (8.2 Å) は、Hisの距離 (4.5 Å) よりも長く、TM1459の安定なタンパク質足場内で目的の配位構造を構築するのに適していると考えられた。
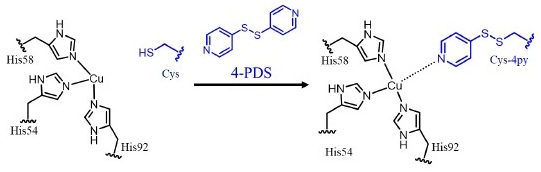
図5 Cys-4pyを形成するための4-PDSによるCysの化学修飾
Cys-4pyの導入に際して、4-PDSとの副反応を避けるため、内在性システイン残基Cys106を欠失させる必要があった。先行研究において、H52A/C106Dがマイケル付加反応において高い収率とエナンチオ選択性を示していたため、この変異体を新たなシステイン残基を導入するための足場として選択した。4-PDSの導入位置として、Ile14、Lys18、Lys24、およびIle49の4アミノ酸残基を選定した。H52Aの結晶構造から、これらの位置のCα原子と銅中心の距離は8.3 Åから13.4 Åと推定された。Cys-4pyの距離 (8.2 Å) を考慮すると、Cys-4pyの配位性窒素原子と銅中心の距離は0.1 Åから5.2 Åの範囲になると見積もられた。調製された変異体タンパク質は、4-PDSとの反応後、ESI-MSにより化学修飾されたことを確認した。ESI-MSの結果は、導入されたほぼすべてのCys残基が4-PDSと反応し、Cys-4pyが生成されたことを示唆している。
次に、それらを用いてニトロメタンのマイケル付加反応を行った。H52A/C106DはS体に対して90%のエナンチオ選択性 (ee) を示した。Cys-4pyをもつI14C-4py、K18C-4py、K24C-4py、I49C-4pyの内、K24C-4pyとI49C-4pyにおいて、エナンチオ選択性の反転が観測でき、特にI49C-4pyはR体をee=71%で生成した。このことからCys49への4py-修飾は、H52A/C106D変異体におけるエナンチオ選択性反転に最も適していることが判明した。49番目の残基が4つの修飾位置の中で、銅中心からCαまでの距離が最も短い (8.3 Å) ために、アザカルコンのSi面側に位置する疎水性ポケットが空間的に充填され、R体に対するエナンチオ選択性を誘発したためと考えられる。
そこで立体選択性の反転を構造から明らかにするため、銅結合I49C-4py/H52A/C106Dの構造を1.07 Åの分解能で決定した (図6A、B)。全体の構造は銅結合H52Aとほぼ同一であり (233 Cα原子のRMSD=0.45 Å)、C106D変異や4py化学修飾によってタンパク質骨格が大きく変化していないことが示された。ただし、ピリジル部分の電子密度は観察されず、熱的に大きく揺らいでいることが示唆された。銅中心の配位構造は、H52Aと同様に、2つの水分子がシス位に位置し、N3O2の四角錐形を完成させるfac-トライアドを示した。このように、Si面側の疎水性ポケットが、導入されたチオピリジンで満たされ、エナンチオ選択性を反転させたことがより明確に示された。
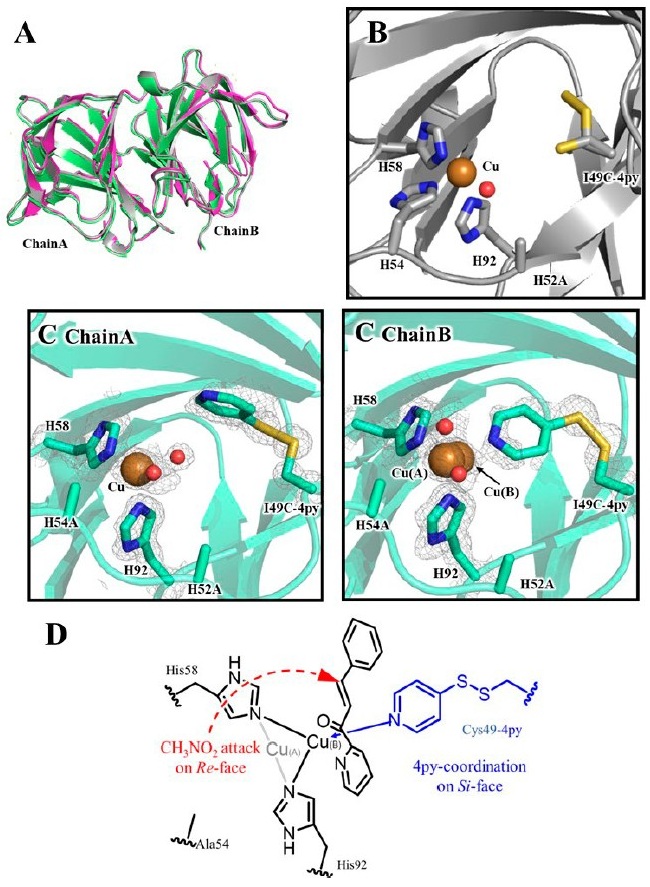

H52Aの活性部位の鏡像対称性を持つ配位構造を構築するため、I49C-4py/H52A/C106DにH54A変異が導入された。これにより調製されたI49C-4py/H52A/H54A/C106Dは、I49C-4py/H52A/C106Dよりも高いR体のエナンチオ選択性 (ee=88% (R) ) を示した。このI49C-4py/H52A/H54A/C106Dの配位構造を確認するため、その銅結合構造を1.08 Åの分解能で決定した (図6A、C)。銅結合I49C-4py/H52A/H54A/C106Dの全体構造は、銅結合H52Aとほぼ同一であり (232 Cα原子のRMSD=0.21 Å)、H54A/C106D二重変異や化学修飾によってもタンパク質骨格に大きな変化がないことが確認された。興味深いことに、I49C-4py/H52A/C106Dとは異なり、この変異体ではピリジル部分の電子密度が観察された。
また結晶構造解析の結果、2量体の各モノマー間で構造に差が見られた。ChainAにおいては、ピリジル部分は銅中心に配位せず、Phe41とのπ–π相互作用によって安定化されていた。そしてHis58、His92、および2つの水分子が銅中心に配位していた。ドッキングシミュレーションにより、この鎖ではRe面側がタンパク質マトリックスによって完全に覆われ、Si面側が溶媒に露出していることが示された。一方でChianBでは、 ピリジル部分が銅中心に配位し、His58、His92、ピリジル基、および2つの水分子が銅中心に配位していた。Si面がCys-4pyによって覆われ、Re面が溶媒に露出していることが示された。さらにドッキングシミュレーションの結果、ChainBにおけるニトロメタンはRe面優先的な攻撃が起こることが示唆された。このことは、Si面側からの攻撃が示唆されていたH52Aの鏡像対称性を持つ配位構造が、エナンチオ選択性の反転をもたらしたことを明確に裏付けた。
最後にCu結合I49C-4py/H52A/H54A/C106Dの金属結合部位では、メジャー種のCu(A)とマイナー種のCu(B)という2つの銅位置が観察され、それぞれの占有率は電子密度から0.51と0.30と推定された。Cu(B)はピリジル基に引き寄せられ、Cu(A)-Cu(B)間の距離は0.86 Åであった。先に示したChainAとChainBの配位構造の違いから、可逆的なピリジンの銅への配位が、銅中心の運動を誘発する可能性が示唆された。この銅の遷移の結果、Cu(B)に結合した2-アザカルコンの位置が疎水性キャビティへと空間的にシフトし、Cu(B)に結合した2-アザカルコンと比較して、Re面側が開放されるとともにSi面側がブロックされると考えられる (図6D)。したがって、この柔軟な銅中心がR体のエナンチオ選択性を向上させたと考察できる。
本研究では、合理的な設計と化学修飾を組み合わせることで、エナンチオ選択性の制御に成功した。さらに、合理的な配位構造の設計に基づき、4-PDSを用いた化学修飾を通して、ニトロメタンの2-アザカルコンへのマイケル付加反応におけるエナンチオ選択性を制御できることを示した。X線結晶構造解析は、Cys-4pyが配位した銅結合部位の構造を明確に決定した。このことから、4py修飾は、従来のタンパク質足場を基盤としたArMにおいて、設計された配位構造を創出するために活用できることが示された。さらに金属の柔軟性により、空間的な遷移を生み出すことで、エナンチオ選択性を向上させることも示唆された。本研究で確立された化学修飾に基づく金属結合部位の構築法は、ArMによって触媒される様々な反応に応用可能であると考えられる。
4.おわりに
以上のようにTM1459タンパク質を用いた立体選択的マイケル付加反応の例について報告した。この2例において選択性を向上させた変異体では、いずれも柔軟な金属中心をもち、主要なCu種とマイナーなCu種が共存するという結果が得られた。これは天然の金属酵素に見られる、金属の移動による活性の制御を、ArMにおいて再現した例となると考えている。このように今回、特殊な金属中心の構造的特徴を得ることができた。得られた知見はArMの反応制御に繋がる新しい戦略の一つとなると期待される。今後は計算手法などを併せて使用することで、アミノ酸残基変異や金属中心の配位挙動や位置遷移を自在に設計することができれば、ArMの適応範囲を更に広げることができると考えられる。
文献
1) Vornholt, T., Leiss-Maier, F., Jeong, W. J., Zeymer, C., Song, W. J., Roelfes, G., Ward, T. R.: Nature Reviews Methods Primers, 4, Article 78 (2024).
2) Dydio, P., Key, H. M., Nazarenko, A., Rha, J. Y. -E., Seyedkazemi, V., Clark, D. S., Hartwig, J. F.: Science, 354, 102 (2016).
3) Key, H. M., Dydio, P., Clark, D. S., Hartwig, J. F.: Nature, 534, 534 (2016).
4) Liang, A. D., Serrano-Plana, J., Peterson, R. L., Ward, T. R.: Acc. Chem. Res., 52, 585 (2019).
5) Waser, V., Mukherjee, M., Tachibana, R., Igareta, N. V., Ward, T. R.: J. Am. Chem. Soc., 145, 14823 (2023).
6) Jeong, W. J., Song, W. J.: Nat. Commun., 13, 6844 (2022).
7) Fujieda, N., Nakano, T., Taniguchi, Y., Ichihashi, H., Sugimoto, H., Morimoto, Y., Nishikawa, Y., Kurisu, G., Itoh, S.: J. Am. Chem. Soc., 139, 5149 (2017).
8) Fujieda, N., Ichihashi, H., Yuasa, M., Nishikawa, Y., Kurisu, G., Itoh, S.: Angew. Chem. Int. Ed., 59, 7717 (2020).
9) Hagedoorn, P. L., Pabst, M., Hanefeld, U.: Applied Microbiology and Biotechnology, 108, Article 391 (2024).
10) Kovalevsky, Y. A., Hanson, L., Fisher, S. Z., Mustyakimov, M., Mason, S. A., Forsyth, V. T., Blakeley, M. P., Keen, D. A., Wagner, T., Carrell, H. L., Katz, A. K., Glusker, J. P., Langan, P.: Structure, 18, 688 (2010).
11) Marsden, S., Wijma, H., Mohr, M., Justo, I., Hagedoorn, P., Laustsen, J., Jeffries, Cy M., Svergun, D., Mestrom, L., McMillan, D., Bento, I., Hanefeld, U.: Angew. Chem. Int. Ed., 61, e202213338 (2022).
12) Matsumoto, R., Yoshioka, S., Yuasa, M., Morita, Y., Kurisu, G., Fujieda, N.: Chem. Sci., 14, 3932 (2023).
13) Podtetenieff, J., Taglieber, A., Bill, E., Reijerse, E. J., Reetz, M. T.: Angew. Chem. Int. Ed., 49 (30), 5151 (2010).
14) Song, W., Jaeseung, Tezcan, F. A.: Science, 346, 1525 (2014).
15) Morita, Y., Kubo, H., Matsumoto, R., Fujieda, N.: Journal of Inorganic Biochemistry, 260, 112694 (2024).
16) Klemencic, E., Brewster, R. C., Ali, H. S., Richardson, J. M., Jarvis, A. G.: Catal. Sci. Technol., 14, 1622 (2024).
17) Lander, A. J., Jin, Y., Luk, L. Y. P.: ChemBioChem, 24, e202200537 (2023).
![]()