�y�g�s�b�N�X�z
���A���^�C��NMR�@�ɂ��APOBEC3G��DNA�f�A�~�l�[�V���������̒�ʉ��
�Ð숟��q
���l�s��@�E������Ȋw
�P�D�͂��߂�
�q�g�Ɖu�s�S�E�C���X (HIV) �͖Ɖu�זE�n�̏h����q�𗘗p���Ď��ȑ��B���J��Ԃ��A��V���Ɖu�s�S�nj�Q (AIDS) �������炷�E�C���X�ŁA2013�N�̐V�KHIV�����Ґ��͂��悻210���l�ł���B���݂̎��Ö@�͂����ɑ��ܕ��p�Ö@�ł��邪�A�d�Ăȕ���p���ܑϐ��ψي��̔��������Ƃ���Ă���B����������HIV�����̊g���h���ɂ́AHIV���̂�W�I�Ƃ����]���̖�܂̊J�������ł͕s�\���ł���A�Ⴆ�Ώh�呤���q����яh����q-HIV�ԑ��ݍ�p��W�I�Ƃ���Ȃǂ̕ψقɑR����V���ȊJ���헪�����߂��Ă���B�����ŁA�ߔN�������ꂽHIV�̑��B�j�Q�������炷�h��̐��̖h��@�\�����ڂ���Ă���B���̈���h��R����APOlipo-protein B mRNA-Editing enzyme Catalyticpolypeptide-like 3G (APOBEC3G; A3G) �^���p�N���ɂ��f�A�~�l�[�V���� (�E�A�~�m��) �����ł���1)�BA3G�́AActivation-Induced cytidine-Deaminase (AID) �ɑ�\�����V�g�V���f�A�~�l�[�X�y�f�̃��`�[�t�z�� (H-X-E-X23-28-P-C-X-C) ��N���[��C���[�ɗL���Ă���B���̂�����C���[�h���C�� (CD2) �݂̂��f�A�~�l�[�X������ێ����Ă���A�h��זE���ŋt�]�ʂ���Đ�����HIV�� (−) DNA���̃V�g�V�����f�A�~�l�[�V�������ăE���V���ɕϊ����邱�Ƃ��ł���B���̕ϊ��́AHIV�� (+) DNA���ł�G����A�ւ̕ψق������炵�A���̌��ʃA�~�m�_�̒u�����~�R�h���̑}���ɔ���HIV �̑����j�Q�ɂȂ���BA3G�̍RHIV �����̍����ł���f�A�~�l�[�V���������@�\�́A�����w�I��@��p���Č�������Ă������ADNA���ɎU�݂���V�g�V���A���z�� (�V�g�V���N���X�^�[) ��N���X�^�[���̃V�g�V���̋�ʂ͍���Ȃ��߁A��p�@���̏ڍׂ͖��𖾂ł������B�{�e�ł́A����A3G�̃f�A�~�l�[�V���������@�\�����A���^�C��NMR�@�ɂ���ĉ𖾂����ߒ����������2)�B
�Q�DAPOBEC3G (A3G) �̍y�f�����̓���
A3G�́A��{��DNA���ɑ��݂���V�g�V���A���z��CCC�̎O�Ԗڂ̃V�g�V�����D��Ńf�A�~�l�[�V�������邱�Ƃ��m���Ă���3)�B�܂��A�����[�����Ƃ�A3G�ɂ��f�A�~�l�[�V���������͈ʒu�ˑ��I�ł���A5’���ɂ��߂��V�g�V���A���z��ɗD��I�ɕψق����������B����ɁA����AFM��u�����G�l���M�[�ړ� (FRET) ���͂Ȃǂɂ���āAA3G�͈�{��DNA�ɔ���ٓI�Ɍ������邽�ߌ����\�����Ɏア���Ƃ�ADNA��𗼕����ɖ�30 nm (69 nucleotides) �قǃX���C�h�������邱�Ƃ�������Ă���4,5)�B���̂悤�ɓ���ȍy�f�����́A�~�J�G���X�E�����e�����ł̉�͂�����ł���B�����ŁA�Ǝ��̔������x�_���f�����\�z���A���A���^�C��NMR�@�Ƒg�ݍ��킹�邱�Ƃɂ���āAA3G�̍y�f�����@�\�𖾂炩�ɂ����B
�\1�@���A���^�C��NMR��p������ƃ^�C���X�P�[��

�R�D���A���^�C��NMR
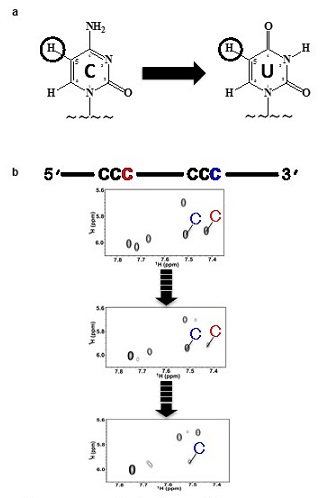
���A���^�C��NMR�@�́A���Ԉˑ��I�ɕω����鉻�w�V�t�g��V�O�i�����x��ǐՂ��邱�ƂŁA��ӁA�|���C���ADNA�C���Ȃǂ̐���������^���p�N���̍\���ω��A�t�H�[���f�B���O���ʓI�ɉ�͂ł�����@�ł���A�y�f�����̒�ʂɂ��K�p�ł���B�����āA�l�X�Ȑ������ۂɑ��郊�A���^�C��NMR�@�̓K�p�Ⴊ����Ă��� (�\1)6)�B���w�V�t�g�̕ω���ǐՂ�����Ƃ��ẮA15N-HSQC (Heteronuclear Single Quantum Coherence spectroscopy) ��NOESY (Nuclear Overhauser SpectroscopY) �X�y�N�g�����o���I�ɑ��肷�邱�Ƃɂ��^���p�N���̃t�H�[���f�B���O�ߒ���͂�����7)�B15N-HSQC�́A���ڌ�������1H�j��15N�j�̑��փX�y�N�g����^����NMR ����@�ŁA�^���p�N�����̎卽�A�~�h��Ƒ����A�~�m��R���̃V�O�i�����I��I�Ɋϑ������BNOESY�́A��ԓI�ɋ߂� (�`5 Å�ȓ�)1H�j���m�̑��ւ����o���鑪��@�ł���B������̑�������̎��̃^���p�N���̍\����Ԃf���č��X�ƕω�����e�V�O�i���̉��w�V�t�g�����A���^�C���Ŋϑ��ł��邽�߁A�t�H�[���f�B���O�ߒ��̉�͂��\�ƂȂ�B�V�O�i�����x��ǐՂ�����Ƃ��ẮAGTPase��GTP��������������GDP�ɕϊ����锽����GTPase��15N-HSQC�X�y�N�g���̌o���I����ʼn�͂��Ă��������8)�B���̂悤�ɁA���A���^�C��NMR�@�́A�K����NMR����@��I�����邱�Ƃɂ���āA�i�s���̗l�X�Ȕ������~�߂邱�ƂȂ��ԗ��I���ڍׂɒǐՂł����@�ł���B�܂��A���q���̕����̗̈�œ����ɋN���锽�����X�ɒǐՂł��邱�Ƃ����_�̈�ł���B�{�����ł́AA3G����{��DNA��̊e���ň����N���������̃f�A�~�l�[�V�����������A1H-1H TOCSY�@ (Totally Correlated SpectroscopY) �ɂ���ĉ�͂��� (�}1b)�BTOCSY�@�Ƃ́A���ډ��w�������Ă���1H�Ԃ̑��փV�O�i�����ϑ������@�ł���A����̎����ł̓V�g�V����5�ʂ�1H��6�ʂ�1H�Ƃ̑��փV�O�i���̌o���ω��ɒ��ڂ��� (�}1a)�B

�\2�@�����ɗp�����j�_�̎��

�S�D���A���^�C��NMR�@�ɂ��A3G�̈ʒu�ˑ��I�ȃf�A�~�l�[�V���������̉��
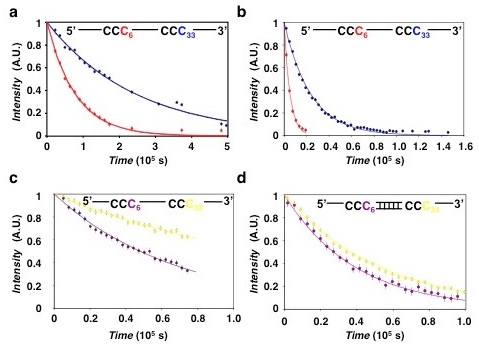
NMR�����ǂɕW�I��{��DNA (�\2) �����AA3G�ɂ�锽���ɔ����ď�������V�g�V���R���̃V�O�i�����x�����A���^�C���ŒǐՂ����B��̓I�ɂ́A�����t��TOCSY�X�y�N�g����A�����肵�A�f�A�~�l�[�V���������ɂ���ď�������V�g�V����5�ʂ�6�ʂ�1H���փV�O�i�����x�����Ԃɑ��ăv���b�g���� (�}1,2)�B�ŏ��ɁA2��CCC���܂ވ�{��DNA (S2ccc) �ɑ���A3G�S����A3G�� CD2�h���C�� (A3G CD2) �̃f�A�~�l�[�V�������������ꂼ��ǐՂ��� (�}2a,b)�B���̌��ʁA�ǂ���̏ꍇ�ł�5’����CCC�ɂ�����f�A�~�l�[�V���������̕����������Ƃ����������B�܂��AA3G CD2�ł�A3G�S���Ɠ��l�Ɉʒu�ˑ��I�ȃf�A�~�l�[�V�����������N�������Ƃ�AA3G CD2��A3G�S�����������y�f�������������Ƃ𖾂炩�ɂ����B���Ȃ킿�AA3G CD2�͈ʒu�ˑ��I�f�A�~�l�[�V�����������N�����̂ɏd�v�ł��邱�Ƃ��������ꂽ�B�����āAA3G ����{��DNA����X���C�h���ăf�A�~�l�[�V�����������N�����Ă��邩�ۂ��ׂ邽�߂ɁA2��CCC��L�����{��DNA���܂ފj�_ (Scccdsccc) ��p���ē��l�̎������s���� (�}2c,d)�BA3G�͓�{��DNA�ɂ͌������Ȃ����߁A��{��DNA��A3G�̃X���C�h��j�Q����B�������ʂ́A�X���C�h���ۂ��N���Ȃ��������Ƃ���������悤�ɁA5’����3’����CCC�ɑ���f�A�~�l�[�V�������x���قړ��������� (�}2d)�B
���̂��Ƃ́A���A���^�C��NMR�@�ł�A3G CD2��DNA����X���C�h���錻�ۂ����o�\�ł��邱�Ƃ������Ă���B����ɁA�قȂ�ʒu��CCC���܂ޓ���������3��ނ̈�{��DNA (S5’CCC�ASmCCC�AS3’CCC) ��p���āA�����ɑ���f�A�~�l�[�V�����̔������x���r�������ʁA5’�� (S5’CCC) >���� (SmCCC) >3’�� (S3’CCC) �̏��ł���A5’���̃V�g�V���A���z��ق�A3G�̍�p���₷�����Ƃ����A���^�C��NMR�@�ɂ���Ċm�F���ꂽ (�}3b)�B


�T�D���f���̍\�z
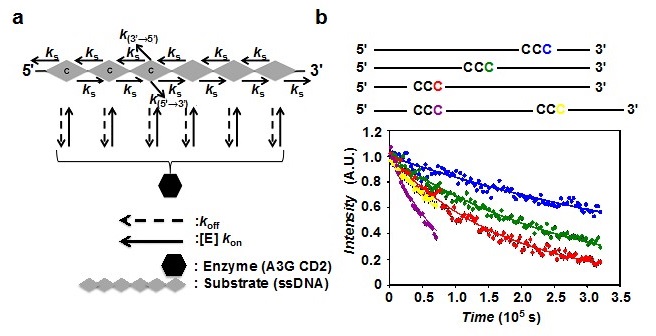
A3G CD2�̈ʒu�ˑ��I�f�A�~�l�[�V���������@�\��������邽�߂ɁA�������x�_���f�����\�z���� (�}3a)�B
���̃��f���́A����@����C�Ɋ�Â����̂ł���B�@�AA3G CD2�́A�ǂ̉���ɂ������������x�萔kon�Ō������A�����𗣌����萔koff�ʼn𗣂���B�A�A�������ɓ������xks��DNA����X���C�h���A��x�X���C�h���n�߂�ƌ�����ς��Ȃ��B�B�A3’����CCC�ɓ��B�����ꍇ�̍y�f����kcat(3’→5’)�ƁA5’����CCC�ɓ��B�����ꍇ�̍y�f����kcat(5’→3’)�Ƃ͈قȂ�BA3G�ƈ�{��DNA�Ƃ̌����\���͖�����Ȃ���A�ȑO�ɕM�҂炪���肵��A3G CD2�̗n�t�\���ɂ͊����|�P�b�g�̕Б��ɕǂ����݂��� (�}4)9,10)�B���̕ǂ��ACCC�𐳊m�Ɋ����|�P�b�g�ɗU�����������S���Ɛ����ł���BA3G CD2�̊����|�P�b�g��CCC���ǂ����z���ĒB���邩�A���B��ɕǂɑ������邩�́A�X���C�h��������Ɉˑ����邱�Ƃ���kcat�̓X���C�h��������ňقȂ�ƍl������ (�}3a)�B�C�A�ʏ�̍y�f�����ł́A�y�f�͐������Ɍ����ł��Ȃ����AA3G�͈�{��DNA�ɔ���ٓI�Ɍ������邽�ߍy�f�ɑ����Ɛ������̘a�͈��Ƃ݂Ȃ���B���̂��߁AA3G�ɂ��f�A�~�l�[�V���������̕ω��ʂ́A��(1)�̂悤�ɕ\�����Ƃ��ł���B����ɁA��q�̉���Ɋ�Â����f����莮������ (�� (2) - (5))�B
![]()
I0�́A����J�n���̃V�O�i�� (�V�g�V����5�ʂ�6�ʂ�1H-1H����) ���x�ł���Akdeami�͌������̃f�A�~�l�[�V�������x��\���A���̂悤�ɋL�q�ł���B
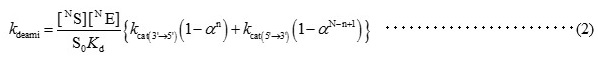
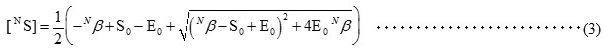
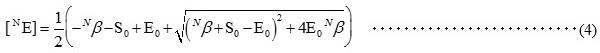
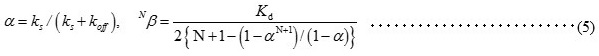
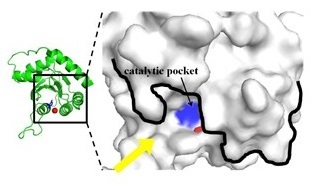
�}4�@A3G CD2�̊����|�P�b�g�\��
N�́A�S���̉���̐���\���An�͔�������V�g�V����3’���[����̈ʒu��\���B[NS]�̓t���[�̊�Z�x�A[NE]�̓t���[�̍y�f�̔Z�x�ł���Bα�AKd (=koff /kon)�Akcat(3’→5’)�Akcat(5’→3’)�́A��������Ɉˑ����Ȃ��p�����[�^�[�ł���B DNA�̍�����A3G��DNA�̃������l�X�ɕς��đ��肵�������^�C��NMR�f�[�^ (S5’CCC�ASmCCC �AS3’CCC��SCCCssCCC���̊eCCC�ɑ��銈��) �̃O���[�o���t�B�b�g���s�������ʁA��L (1) ���ɗǂ��t�B�b�g���Akcat(3’→5’) ��68 s-1�Akcat(5’→3’) ��14 s-1�Ƃ������ʂ�����ꂽ11)�B���Ȃ킿�AA3G�ɂ��f�A�~�l�[�V�����������ʒu�ˑ���������������2�̈قȂ�kcat�ɂ����̂ł���AA3G��CCC�ɑ���3’������ڋ߂�������A5’������̐ڋ߂����f�A�~�l�[�V�����������N����₷�����Ƃ������ꂽ�BA3G�́A��{��DNA�̑S�̈�ɋϓ��Ɍ���������A�ǂ���̕����ɂ������m���ŃX���C�h����B���̏ꍇ�A5’���Ɉʒu����CCC�̕����AA3G��3’������̐ڋߕp�x�������Ȃ�B�X��CCC�ɑ���A3G�̊����͓����ł���Ȃ���A3’������̐ڋߕp�x��CCC�̈ʒu�ɂ���ĈقȂ邽�߁A���ʂƂ���A3G��5’����CCC�ɑ��Ă�荂�p�x�ɕψق�����̂ł���B
�U�D������
�f�A�~�l�[�V���������̃��A���^�C��NMR�f�[�^��V�K�ɍ\�z�����������x�_���f���ɑ����ĉ�͂��邱�Ƃɂ���āAA3G�̈ʒu�ˑ��I�f�A�~�l�[�V���������@�\�𖾂炩�ɂ����B�f�A�~�l�[�V���������̈ʒu�ˑ����́AA3G���W�I�V�g�V���ɐڋ߂�������ɂ���ĈقȂ�y�f����kcat���������ƂɋN������Ɣ������� (�}5)�B
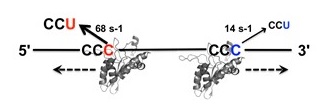
�}5�@A3GCD2�̈ʒu�ˑ��I�f�A�~�l�[�V���������@�\
APOBEC�t�@�~���[�ɂ��f�A�~�l�[�V�����������ADNA�̒E���`�����ߒ��Ɋ֗^���邱�Ƃ����炩�ɂ������B�܂��A���̍y�f�ɂ����DNA���̃V�g�V�������`�����E�q�h���L�V���`�����ȂǗl�X�ȏC�����A�G�s�W�F�l�e�B�b�N���ߋ@�\�̈�U��S���Ƃ̕�����Ă���B����琶�����ۂ̍����𐬂��y�f�����̏ڍׂȔ����@�\�͉𖾂���Ă��炸�A����Љ�����A���^�C��NMR�@����g���ĉ�͂�i�߂Ă���Ƃ���ł���B
�ӎ�
�{�e�ŏq�ׂ������́A���s��w�G�l���M�[���H�w�������̕Е����l�����Ɖi�c���y�����A����ы��s��w��w�@�H�w�����ȕ��q�H�w��U�̐��������y�����̂��x���ɂ�芮�����邱�Ƃ��ł��܂����B���̏�����肵�Ċ��ӂ̈ӂ�\���܂��B
����
1) Sheehy, A. M., Gaddis, N. C., Choi, J. D., Malim, M. H.: Nature, 418, 646 (2002).
2) Furukawa, A., Sugase, K., Morishita, R., Nagata, T., Kodaki, T., Takaori-Kondo, A., Ryo, A., Katahira, M.: Angew. Chem. Int. Ed., 53, 2349 (2014).
3) Yu, Q., Konig, R., Pillai, S., Chiles, K., Kearney, M., Palmer, S., Richman, D., Coffin, J. M., Landau, N. R.: Nat. Struct. Mol. Biol., 11, 435 (2011).
4) Shlyakhtenko, S., Lushnikov, A. Y., Miyagi, A., Li, M., Harris, R. S., Lyubchenko, Y. L.: Biochemistry, 51, 6434 (2012).
5) Senavirathne, G., Jaszczur, M., Auerbach, P. A., Upton, T. G., Chelico, L., Goodman, M. F., Rueda, D.: J. Biol. Chem., 287, 15826 (2012).
6) Smith, M. J., Marshall, C. B., Theillet, F. X., Binolfi, A., Selenko, P., Ikura M.: Curr. Opin. Struct. Biol., 32, 39 (2015).
7) Mizuguchi, M., Kroon, G. J., Wright, P. E., Dyson, H. J.: J. Mol. Biol., 328, 1161 (2003).
8) Mazhab-Jafari, M. T., Marshall, C. B., Smith, M., Gasmi-Seabrook, G. M. C., Stambolic, V., Rottapel, R., Neel, B. G., Ikura, M.: J. Biol. Chem., 285, 5132 (2010).
9) Holden, L. G., Prochnow, C., Chang, Y. P., Bransteitter, R., Chelico, L., Sen, U., Stevens, R. C., Goodman, M. F., Chen, X. S.: Nature, 456, 121 (2008).
10) Furukawa, A., Nagata, T., Matsugami, A., Habu, Y., Sugiyama, R., Hayashi, F., Kobayashi, N., Yokoyama, S., Takaku, H., Katahira M.: EMBO J., 28, 440 (2009).
11) Sugase, K., Konuma, T., Lansing, J. C., Wright, P. E.: J. Biomol. NMR, 56, 275. (2013).
![]() �@
�@