【トピックス】
酵素反応を鍵とした非天然型 (非タンパク質性) アミノ酸の製造
安田磨理
エーピーアイ コーポレーション
1.はじめに
非天然型 (非タンパク質性) アミノ酸は様々な医薬中間体としての需要があり、薬理活性の向上、安定性向上、膜透過性向上などを目的に用いられ1)、特にペプチド型の医薬品の増加に従い、その重要性は益々高まっている。これら特殊なアミノ酸需要を背景に、多くの研究グループが新しい化学的アミノ酸製造方法や酵素的合成を研究してきた。一般的に酵素法は、立体選択性、条件の穏和さ、環境負荷などの点で優れているが、反応基質が限定されるなどの問題点もある。
筆者らが所属する三菱化学グループは、長年化学法と酵素法を組み合わせた効率的なプロセスによる医薬中間体の製造を行ってきた2)。その結果、豊富な微生物ライブラリーおよび酵素ライブラリーを保有している。これらの資産を生かし、かつ、酵素の問題点を合成研究者と共に解決して、新規なアミノ酸製造法を開発してきた。ここでは、筆者らの独自酵素反応を鍵とした非天然型 (非タンパク質性) アミノ酸の製造例をいくつか紹介したい。
2.N-メチルアミノ酸 (NMA) の合成
NMAはdolastatins3) やdidemnin4) といった生理活性のある化合物の構成アミノ酸として天然界にも存在し、NMAを含むペプチド型医薬品は種々開発研究が行われている5)。
NMAの合成法はいくつか知られており、総説も発表されている6)。図1(1) に示すように光学活性α-ブロモカルボン酸の求核置換反応によりN-メチルアラニン、N-メチルロイシン、N-メチルフェニルアラニンが合成されている7)。図1(2)〜(5) ではいずれも還元的メチル化の例を示している。光学活性アミノ酸を一旦N-ベンジル化したのち、水素化で目的物を得る方法8)(図1(2))、光学活性アミノ酸の還元的メチル化をホルムアルデヒドと亜鉛を用いて行う方法 (図1(3))、光学活性なα-アジドカルボン酸を原料に用いる方法 (図1(4))、オキザゾリジノンを経る方法 (図1(5)) が知られている。しかし、いずれも光学活性な原料が必要であることや、多段階反応、ラセミ化の問題などがあり、工業的に優れた方法は知られていなかった。
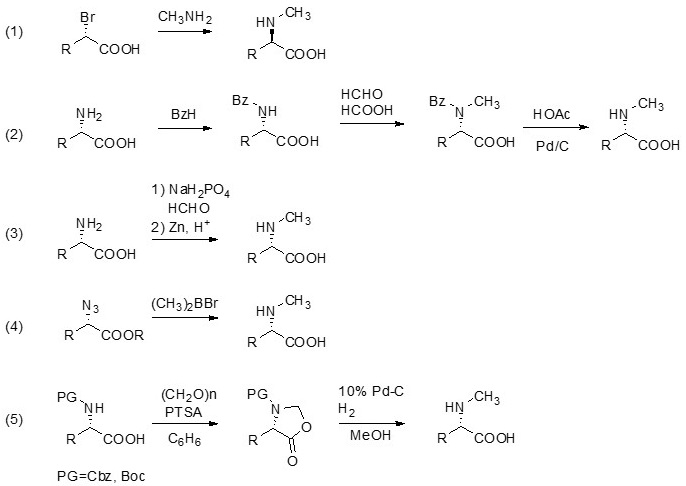
図1 様々なN-NMAの合成方法
筆者らは、Vertex社が開発しているVX-853 (Timcodar dimesilate) の中間体として需要のあるN-メチルフェニルアラニン誘導体 (図2) の効率的な合成方法の検討を開始した。初期にはN-メチルヒダントインの酵素的加水分解などを検討していたが、所望の反応を示す酵素を得ることができなかった。そこで、α-ケト酸に酵素で直接N-メチル化ができないかと考え、微生物のスクリーニングを開始した。当初は突拍子もないアイディアに思われたものの、実際検討してみるといくつかの微生物で活性が確認された。
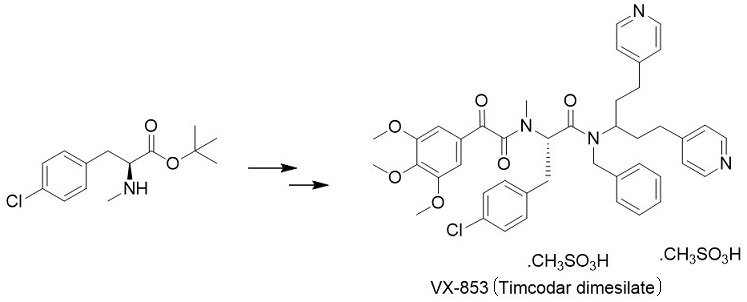
図2 VX-853とその中間体
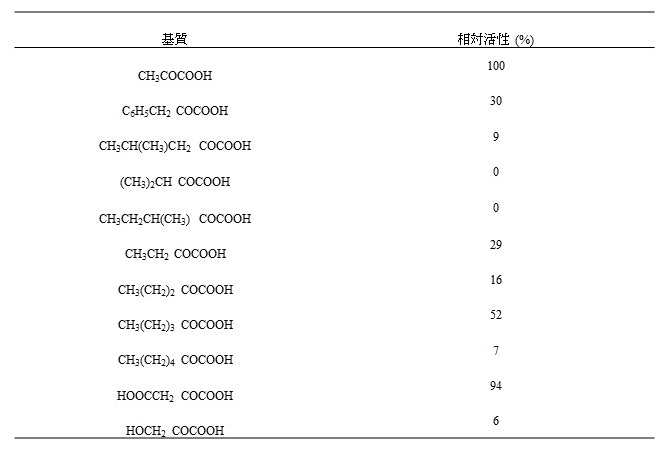
共同研究先の京都大学にてPseudomonas putida ATCC12633より本酵素が精製され、N-methyl-L-amino acid dehydrogenase(NMAADH) と命名された。遺伝子クローニングと大腸菌での酵素高発現にも成功した9)。本酵素は酸化還元酵素の一種で補酵素としてNADPHを必要とする。酵素の諸性質を調べると、アンモニアは基質として認識しないことが判明した。ケト酸類の基質特異性は、表1に示す。
表1 NMAADHの基質特異性
次に、NMAADHを用いての物質生産を検討した。工業的生産には補酵素NADPHの再生系を組み合わせる必要がある。大腸菌内で本酵素とBacillus subtilis由来のglucose dehydrogenaseとを共発現させることにより、N-methyl-L-phenylalanine10) およびN-methyl-L-alanineの効率的な生産方法を確立した。いずれも工業化可能な高濃度生産性を達成した。
基質のα-ケト酸が工業的に入手できない場合に備え、安価なラセミアミノ酸から酵素的にα-ケト酸を生成させ、続けてNMAADHによるN-メチル化を行うというカスケード反応も開発した。図3には、例としてD-アミノ酸からNMAの合成ルートを示してある。
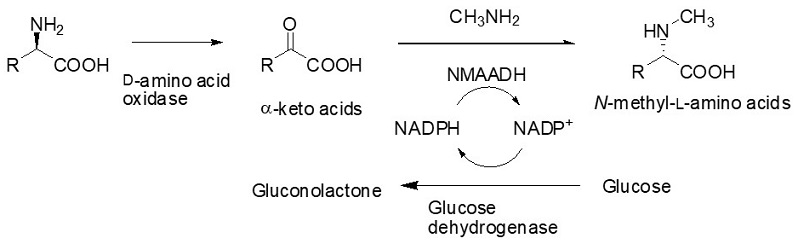
図3 D-アミノ酸からのNMA合成
3.環状アミノ酸 (CAA) の合成
CAAは、ピペコリン酸に代表されるように様々な医薬品の原料や中間体としての需要がある11, 12)。様々な合成研究例が報告されているが13-16)、いずれも多段階の反応であり工業的には適さない方法であった。
NMAADHの基質特異性をさらに検討する中で、Δ1-ピペリジン-2-カルボン酸に対して最も活性を示すことが判明し、本酵素は本来D-リジンの代謝系に関与する酵素であり、イミンの還元を行っていることが分かった。リジンのようなα,ω-ジアミノ酸 (DAA) を原料にアミノ酸オキシダーゼやトランスアミナーゼの作用によりα-ケト-ω-アミノ酸を生成させると、自然環化し環状のイミノ酸が容易に生成される。この反応とNMAADHの反応をカップリングさせることにより、DAAからのCAA生成法を確立することができた。我々の方法の応用性を検討すべく様々なDAAを検討した。図4に反応式と検討基質を示す。何れの基質においても対応するCAAが確認された17)。
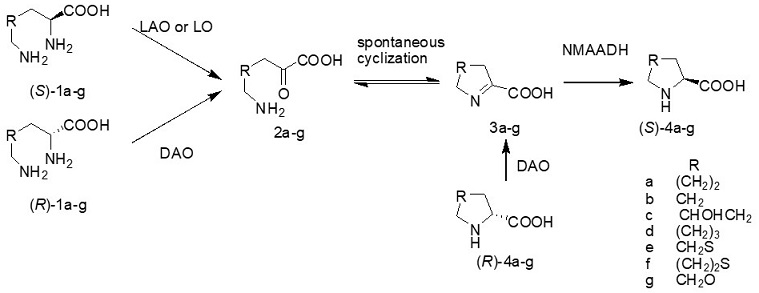
図4 D-DAAおよびL-DAAからのCAAの生成ルート
4.β-ヒドロキシアミノ酸 (bHAA)
bHAAは、セリンやスレオニンに代表されるように天然界にも多く存在するアミノ酸群である。開発薬の抗ウイルス剤ONO-4128には、非天然型のbHAAである (2R,3R)-2-アミノ-3-ヒドロキシ-3-シクロヘキシルプロピオン酸 (ACH) が重要中間体として用いられている (図5)18)。
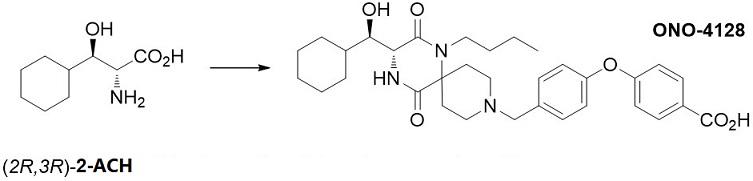
図5 ONO-4128とその中間体
bHAAには2つの隣り合う不斉炭素があり4つの立体異性体が存在するため、目的とする立体異性体を得るために多くのグループが開発研究を行ってきた。光学活性なエポキシ誘導体を、光学分割の擬似移動床や酵素を用いた分割法により取得し目的化合物を合成する方法が報告されているが19)、いずれも原料の50%以上を廃棄する製法である。収率のよい方法として不斉アルドール法も種々報告されている。化学的な不斉アルドール反応ではN-保護のグリシン誘導体および不斉補助基が必要であるが、酵素であれば直接的な不斉アルドール反応が可能である。Steinreiberらはスレオニンアルドラーゼでの反応を検討し、良好な立体選択性を示したが、ジアステレオマー選択性は不十分であった。また、シャープレス酸化による製造法も西山らにより検討され、良好な反応性を示したが、多段階の反応が必要であった。
筆者らは独自のカルボニル還元酵素ライブラリーを有しており、bHAA製法の鍵反応としてカルボニル還元が利用できないかと考えた。α-アミノ-β-ケトエステルは中性域の水溶液中でもラセミ化するため、カルボニル還元酵素が所望の光学異性体のみを基質として認識し、かつ、anti選択的に還元することができれば動的速度論分割 (DKR) となり理論収率100%の製法となる。有機合成では、既に牧野らがRu-BINAP触媒によるDKRでの製造に成功していた20)。ただし、この方法は光学選択性が十分ではない。
筆者らは所望の反応を示す酵素をカルボニル還元酵素ライブラリーから探索することとしたが、この時に基質のデザインを如何にするかが問題となった。最終的にコスト競争力のある製造ルートが必要であり、基質製造およびバイオ反応後工程のコストを意識しながら、かつ酵素が認識しやすい基質デザイン検討を心がけた。合成研究員が数 mgから数十 mgの基質を合成し、それを直ちにバイオ研究員が評価することを繰り返し、基質を検討した。カルボン酸エステルは酵素反応の研究例の多いエチルエステルに絞り、アミノ基の保護基を種々検討した。最初の検討基質としては、図6に示すようにアミノ基をベンジル基で保護した基質を用いた。本ルートのバイオ不斉還元が通れば、安価な原料から4工程で製造できる競争力のあるルート構築が可能となる。しかしながら、本基質にはどの酵素も全く反応性を示さなかった。カルボニル還元酵素による類似の反応例を調べると、アミノ基の保護にはアセチル基などしか知られておらず、大きな保護基での不斉還元の例は全くなかった。そこで次に保護基のない基質で検討したが、反応終了時のサンプルを分析してみると、基質は消失していたが目的生産物は全く得られなかった。
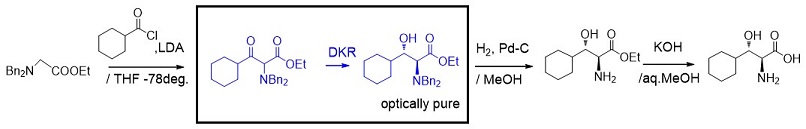
図6 N-ジベンジル基での製造ルートアイディア
次に、N-Boc保護基で検討すると保有酵素ライブラリーのうち2つの酵素が反応性を示し、1つの酵素 (MCRC4) ではジアステレオ選択性99%de以上、光学純度も99%ee以上という極めて高い還元体が生成された。しかしながら、絶対配置を確認すると目的物とは全く逆の立体である (2S, 3S)-ACHと判明した (図7)。ここまでの結果を表2にまとめる。
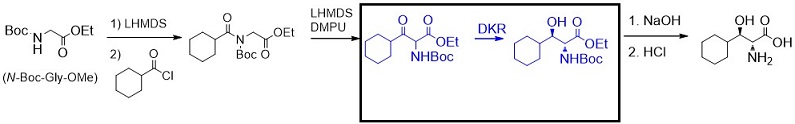
図7 N-Boc保護基での製造ルートアイディア
表2 酵素的DKRにおける基質アミノ基の保護基の影響 (1)
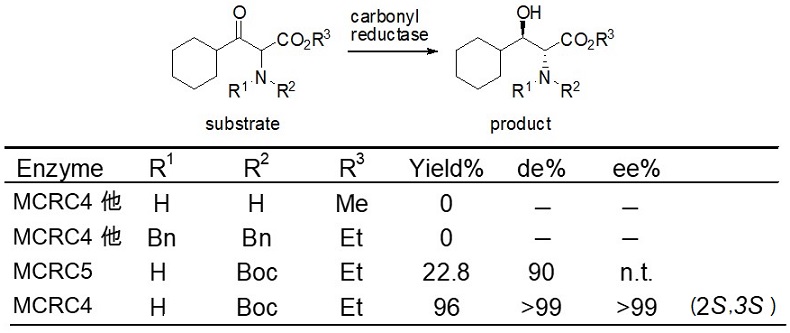
最終的には基質アミノ基の保護基をN-ベンゾイル基にして、酵素MCRC5を用いたときに目的物 (2R, 3R)-ACHが確認できた。しかしながら、この基質を用いると基質のエステル基がホスト大腸菌の持つエステラーゼで加水分解され、続けて脱炭酸が起こり、収率が大きく低下する問題が発生した。この脱炭酸体も同じカルボニル還元酵素で還元され、副生成物として検出された。また、ジアステレオ選択性も十分ではなかった。そこで、種々のエステル基を検討した結果、イソプロピル基を用いたときにジアステレオ選択性が向上し、98%de以上となったが (表3)、エステル基の加水分解を防ぐことはできなかった。また、当時は生産性0.1 g/L以下という極めて低い成績であったことから、さらによりよい結果を示す基質を求め検討を続けた。その結果を表4に示す。しかし、残念ながらN-ベンゾイル体を上回るものはなかった。
表3 酵素的DKRにおける基質エステル基の影響
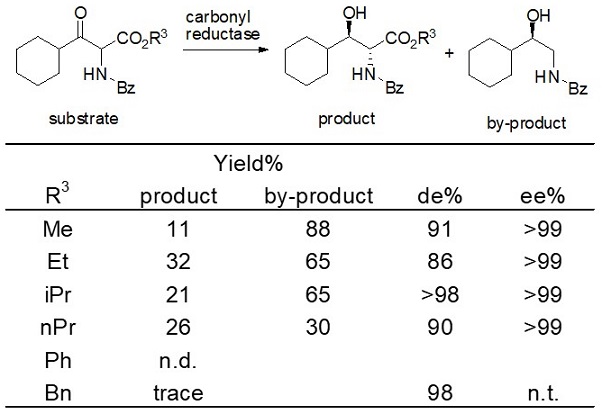
表4 酵素的DKRにおける基質アミノ基の保護基の影響 (2)
最適な酵素を探すために微生物のスクリーニングも行った。多くの微生物で還元活性が確認されたものの、MCRC5酵素を組換えた大腸菌で示された生産性を上回るものはなかった。
還元反応が先に進行すれば、還元体が加水分解されることはない。そこで、反応速度を上げ収率を向上させるため、MCRC5酵素遺伝子にerror prone PCRによるランダム変異導入を行い、directed evolutionによる活性向上を試みた。この検討で鍵となるのは、所望の変異体スクリーニングにおいて、エラーが少なくかつスループットの高いスクリーニング系の構築である。分析項目としては、生産物濃度、ジアステレオ選択性、エナンチオ選択性と多いため、HPLCの高速分析を検討し、ショートカラムなどを用いることで1日に約400株の変異体をスクリーニングすることができた。変異3世代目では、元の酵素活性を大きく上回ることができた (図8)。その結果、最終的には十分工業化ができる反応速度、収率、生産物濃度を達成した。
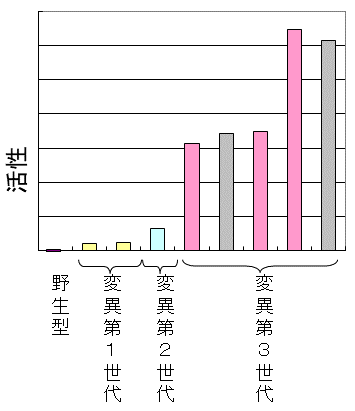
図8 MCRC5酵素のDirected evolutionの結果
一方、合成研究者はさらなる基質合成の効率化を検討し、最終的には安価な原料からワンポットでの基質調製が可能となった。筆者らの最終製造ルートを図9に示す。
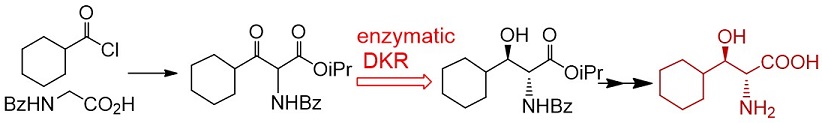
図9 (2R, 3R)-2-ACHの最終製造ルート
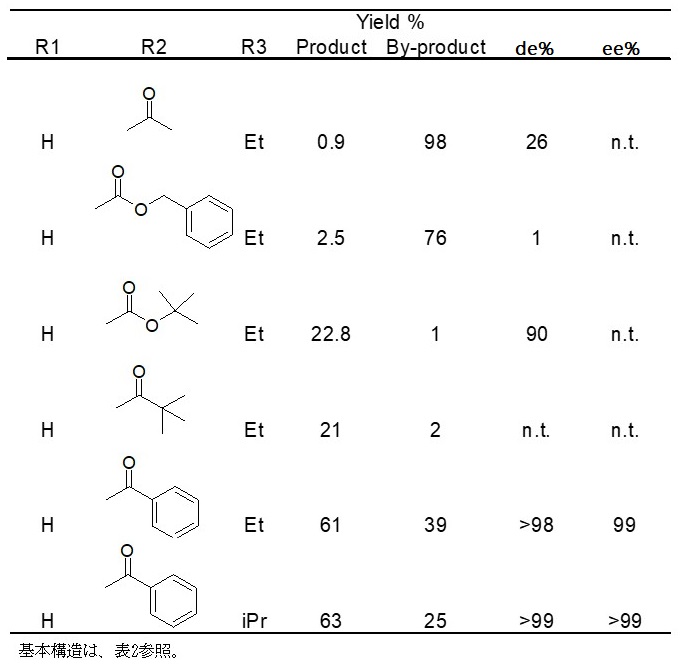
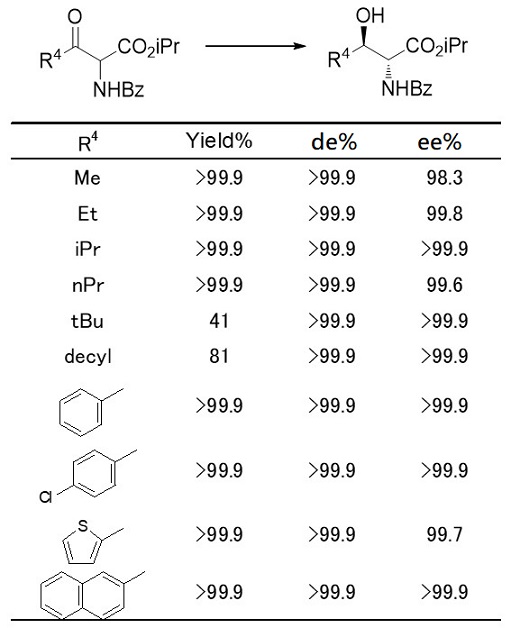
本ルートの応用範囲を確認すべくMCRC5の変異酵素の基質特異性を調べると、シクロヘキシル基以外の多くの基質を認識し、極めて光学純度およびジアステレオ選択性高く還元することがわかった (表5)。野生型酵素は他のアルキル基に比べ、シクロヘキシル基やtert-ブチル基など嵩高いものは、基質として非常に反応性に乏しく、本酵素変異体がこれらの基質に対しても十分反応出来るように改良されたことは、酵素の大きな可能性を示すものである。
表5 酵素的DKRにおける基質特異性の検討
5.おわりに
現在も非天然型アミノ酸の需要は伸びており、我々もさらに新しいターゲット化合物に対して新しい製造ルートを開発すべく日々検討を続けている。近年多くの酵素が市販され、酵素反応の応用例の多くが公知になる中で、競争力のある新しいプロセスを開発するためには、以前にも増して合成とバイオ研究者の知恵を結集して取り組まなければならない。この点、日本は両分野で優れた大学や企業が多くあることから、適切な協奏体制を築くことで世界をリードできる位置にある。多くの研究者が切磋琢磨し、素晴らしい技術で世界の競争に勝ち、日本の存在感を示したいと切に願っている。我々エーピーアイコーポレーションもその一角を担えるよう、新しいプロセス開発に挑戦し続けている。
謝辞
NMAおよびCAAの共同研究で多大なご尽力を頂きました京都大学江崎信芳先生、立命館大学三原久明先生、高知大学村松久司先生、およびbHAAを使用したカルボニル還元酵素を発見して下さった北海道大学和田大先生に深く感謝致します。また、本研究に協力頂いた (株) 三菱化学科学技術研究センターの多くの研究者の方々に感謝します。
文献
1) Rabasseda,
X., Martel, A. M., Castaner, M. J.: Drugs Fut., 22, 371 (1997).
2) 上田 誠:BIOINDUSTRY 7月号, 70 (2008).
3) Rinehart, K. L. Jr., Gloer, J. B., Cook, J. C. Jr.: J. Am. Chem. Soc., 103 1857 (1981).
4) Pettit, G. R., Kamano, Y., Herald, C. L., Fujii, Y., Kizu, H., Boyd, R. R., Boettner, F. E., Doubek, D. L., Schmidt, J. M., Chapuis, J.-C., Michel, C.: Tetrahedron, 49, 9151 (1993).
5) Maes, V., Garayoa, E. G., Bla¨uenstein, P., Tourwe, D.: J. Med. Chem., 49, 1833 (2006).
6) Aurelio, L., Brownlee, R. T. C., Hughes, A. B.: Chem. Rev., 104, 5823 (2004).
7) Fischer, E., Mechel, L. V.: Chem. Ber., 49, 1355 (1916).
8) Quitt, P., Hellerbach, J., Vogler, K.: Helv. Chim. Acta, 46, 327 (1963).
9) Mihara, H., Kakutani, R., Muramatsu, H., Yasuda, M., Ueda, M., Kurihara, T., Esaki, N.: FEBS J., 272, 1117 (2005).
10) Muramatsu, H., Mihara, H., Kakutani, R., Yasuda, M., Ueda, M., Kurihara, T., Esaki, N.: Tetrahedron Asymm., 15, 2841 (2004).
11) Lin, L. S., Lanza, T. Jr., McCauley, E.: Bioorg. Med. Chem. Lett., 12, 133 (2002).
12) Nakatsuka, M., Ragan, J. A., Sammakia, T., Smith, D. B., Uehling, D. E., Schreiber, S. L.: J. Am. Chem. Soc., 112, 5583 (1990).
13) Ohtani, B., Tsuru, S., Nishimoto, S., Kagiya, T., Izawa, K.: J. Org. Chem., 55, 5551 (1990).
14) Shiraiwa, T., Tadokoro, K., Tanaka, H., Nanba, K.,Yokono, N., Shibazaki, K., Kubo, M., Kurokawa, H.: Biosci. Biotech. Biochem., 62, 2382 (1998).
15) Kogami, Y., Okawa, K.: Bull. Chem. Soc. Jpn., 60, 2963 (1987).
16) Seebach, D., Dziadulewicz, E., Behrendt, L., Cantoreggi, S., Fitzi, R.: Liebigs Ann. Chem., 1215 (1989).
17) Yasuda, M., Ueda, M., Muramatsu, H., Mihara, H., Esaki, N.: Tetrahedron Asymm., 17, 1775 (2006).
18) 高岡義和, 西澤玲奈, 柴山史朗, 佐川健二, 松尾政芳: WO 2004/26873
19) 田中洋己, 西川和良, はま嵜亮太, 織田博史, 岡野俊紀, 中島賢則: WO 2005/040099
20) Makino, K., Goto, T., Hiroki, Y., Hamada, Y.: Angew. Chem. Int. Ed., 43, 882 (2004).
![]()