【トピックス】
結晶構造を基盤とした植物ポリケタイド合成酵素の触媒機能の拡張
森田洋行
東大院・薬
1.はじめに
医薬資源として重要な天然物の基本骨格を構築する二次代謝酵素の中には、活性部位の微妙な構造の違いにより、基質特異性や反応様式が大きく変化するものがあり、これが天然物の多様性を生み出す大きな要因になっている。また、一般に、酵素の基質特異性は厳密で自由度が低いものとされているが、これらの中には、それ自体、広範な基質特異性や触媒ポテンシャルを有するものもある。こうした二次代謝酵素が示す広範な基質特異性と触媒ポテンシャルを活用・改変することにより、効率的な物質生産が可能になる。本稿では、植物由来Ⅲ型ポリケタイド合成酵素 (PKS) を取り上げる1,2)。これらは、反応の立体化学が厳密に制御された「精巧な酵素システム」であるとは言い難く、むしろ単純なアシル基転移の繰り返しによる「炭素鎖伸長マシン」と捉えることができる。筆者らは、酵素のX線結晶構造解析の結果から、基質及び生成物特異性を決定する酵素活性中心構造を解明し、さらに合理的な変異の導入により、C2単位縮合数の拡大と新たな芳香環縮合系の構築など、これまで困難とされてきた酵素触媒機能の操作にも展望を開きつつあるので、以下に紹介したい。
2.植物ポリフェノールの基本骨格を構築するⅢ型PKSについて
植物由来Ⅲ型PKSは、分子量約4万のサブユニットからなる、ケト合成酵素 (KS) のみで構成されたホモダイマー酵素であり、フラボノイドやレスベラトロールなど、植物が生産するポリフェノール類の基本骨格を構築する酵素群である1,2)。例えば、植物に普遍的に存在するフラボノイドの骨格を構築するカルコン合成酵素 (CHS) は、1分子のクマロイルCoAを開始基質として、3分子のマロニルCoAに由来するC2単位を順次縮合の後、クライゼン型の閉環反応が進行して、芳香環を形成し、ナリンゲニンカルコンを生成する (図1)。これ以外にも、ブドウなど限られた植物に分布するスチルベン合成酵素 (STS) やホップのバレロフェノン合成酵素 (VPS) など、様々な機能を有するⅢ型PKSが知られている (図1)。このような分子多様性を生み出す要因として、酵素反応の開始基質、ポリケタイド鎖長を決定するマロニルCoAの縮合回数、そして最終的な閉環反応様式の違いが挙げられる。
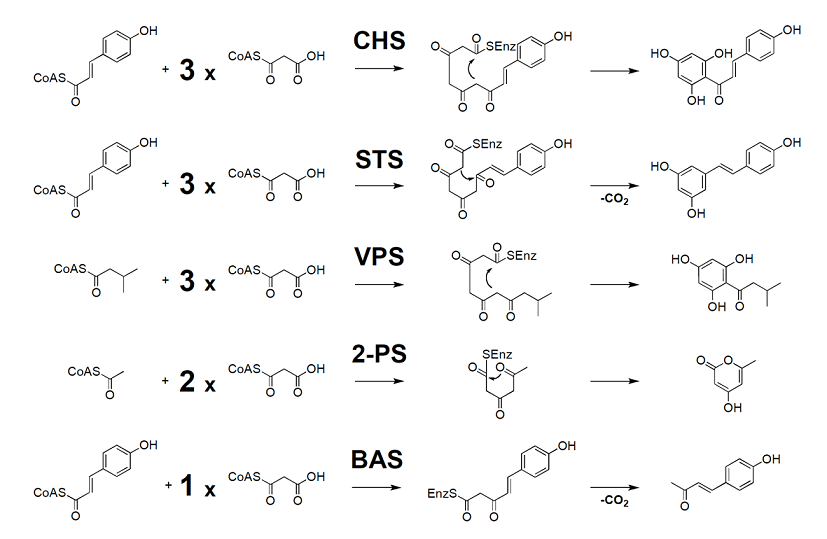

Ⅲ型PKSの酵素反応は、単量体それぞれのほぼ中央部に一つずつ存在する活性中心キャビティに、アシル基が取り込まれることによって開始する。これにより、隣接するHis残基との水素結合により安定化された、活性中心Cys残基のチオレートアニオンが、開始基質のカルボニル炭素を求核攻撃する。次いでこのCys残基を起点として、伸長基質マロニルCoAの脱炭酸を伴ったアシル基の転移を繰り返し、即ち、炭素鎖伸長の度にいったん形成されたチオエステル結合が開裂して、そこに新たなC2単位の挿入を繰り返し、最後にβ-ポリケトメチレン中間体がクライゼンあるいはアルドール型の縮合により閉環して、酵素反応を終了する。この間、HisとAsn残基は、オキシアニオン・ホールを形成し、マイナスに荷電した遷移状態中間体を安定化することにより、マロニルCoAの脱炭酸とその結果生じるエノラートアニオンの求核攻撃を促進する。このCys-His-Asnからなる活性中心触媒残基は、機能の異なる全てのⅢ型ポリケタイド合成酵素において保存されており、同一の反応機構でポリケタイド鎖伸長反応が進行するものと考えられている。一方、既にCHSをはじめとして、7種類の酵素のX線結晶構造解析がなされており、その結果、これら酵素の機能多様性は、主として活性中心キャビティの大きさと形状によって決定されることが示唆されている1-4)。
3.キダチアロエ由来新規Ⅲ型PKS
薬用植物キダチアロエ (Aloe arborescens) から得られたペンタケタイドクロモン合成酵素 (PCS) とオクタケタイド合成酵素 (OKS) は、これまでに例のない全く新しいタイプのⅢ型PKSである5,6)。互いに微妙に異なるアミノ酸配列を有するこの2つの酵素は、植物に普遍的に存在するCHSとはアミノ酸レベルで約60%の配列相同性を示すものの、CHSのようにナリンゲニンカルコン合成能は示さず、それぞれ5分子あるいは8分子のマロニルCoAを直接縮合して芳香族ポリケタイドを生成する (図2)。PCSが産生するクロモンは、抗喘息・抗アレルギー薬のリード化合物として有名なケーリンなど生理活性フロクロモンの生合成前駆体となる。一方、OKSが産生するSEK4/SEK4bは、放線菌のⅡ型のminimal PKS (還元酵素などのサブユニットを欠落させたⅡ型PKSの最小単位の酵素複合体) が産生する代謝産物として知られてはいるものの、Ⅲ型酵素による、マロニルCoA8分子の縮合はこれが最初であり、最多であった。
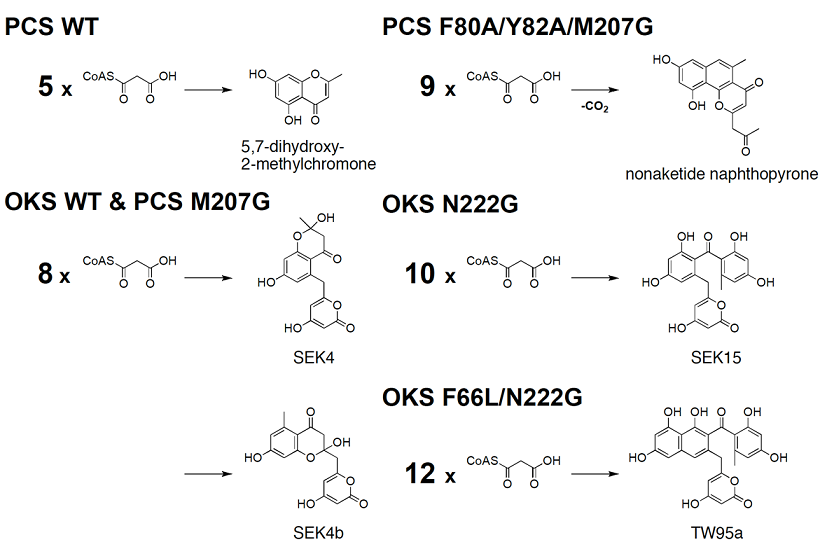

アミノ酸レベルで互いに92%という非常に高い配列相同性を示すこの2つの酵素が、このように全く異なる生成物を与えるのは何故か、マロニルCoAの縮合回数の違いを決定する要因は何か? これら2つの酵素においては、CHSの触媒残基Cys164,His303,Asn336がすべて保存されている一方で、活性中心キャビティを構成するアミノ酸残基Thr197,Gly256,Ser338が、PCSではMet207/Leu266/Val351、OKSではGly207/Leu266/Val351に置換されているのが特徴的である。そこで筆者らは、CHSのThr197残基が、PCSにおいてはMet207に、また、OKSにおいてはGly207に置換されていることに着目し、この残基の部位特異的変異酵素を作成して酵素活性に及ぼす影響を調べた5)。その結果、本来5分子のマロニルCoAを縮合するPCSのM207G置換体では、酵素活性が劇的に変化してマロニルCoA8分子の縮合により、OKSの生成物であるSEK4/SEK4bを生成することを見出した (図2)。
4.X線結晶構造解析に基づくマロニルCoA縮合数の拡張
そこで、PCS野生型、および、そのM207G変異型酵素について、1.6 Åの分解能で結晶構造を解明した7)。まず、アミノ酸レベルで互いに60%の相同性を示すCHSの結晶構造との比較により、両酵素はタンパク全体ではほぼ同一のフォールディングを共有すること、しかも驚くべきことに、その基質特異性と生成物特異性は明らかに異なるものの、活性中心を構成するほとんどのアミノ酸残基を見事に重ねることが可能であった。一方、活性中心キャビティの大きさはPCSの方が明らかに小さく、しかもCHSの活性中心キャビティに見いだされたクマロイル結合ポケットが、PCSの活性中心キャビティから欠損していることが示された (図3)。次に、野生型PCSとM207G変異型酵素の構造の比較により、Met207をGlyに置換することで、それまで後方にもともと埋もれていた新規ポケットが、M207G変異酵素のキャビティの一部となり、その大きさが劇的に変化することが示された (図3)。
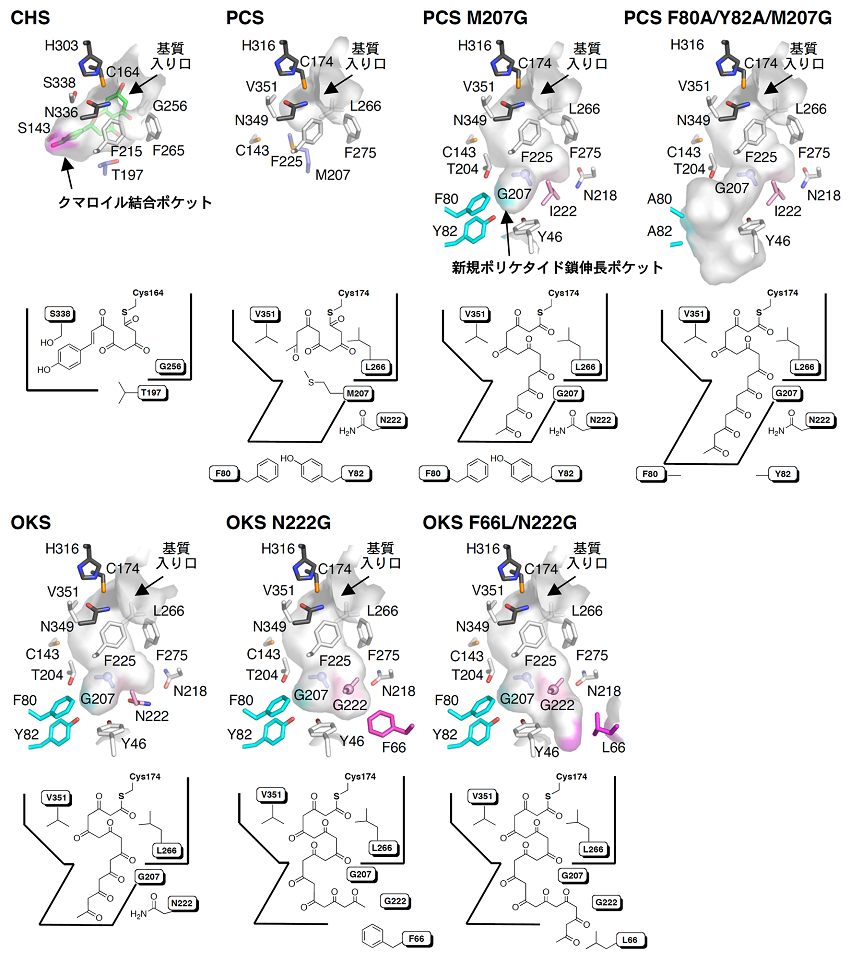

それでは新たに出現したポケットをさらに掘り進めてキャビティを広げてやるとどうなるのか? 上述した解析の結果は、点変異M207Gの導入により、新規ポケットの入り口が開いて、これによりポリケタイド鎖の伸長がさらに進行して、5分子の代わりに8分子のマロニルCoAの縮合が進行することを示しているように見える (図3)。また、PCSにおいては、STSにみられるような閉環反応の制御に重要とされる、Ser338残基近傍の水分子を介した水素結合のネットワーク8) は観察されず、単に活性中心キャビティの大きさと形状の変化によって生成物の特異性が決定されるものと考えられた。そこで筆者らは、ポケットの底面を形成するPhe80, Tyr82の2つの芳香族残基を、同時にAlaで置換したF80A/Y82A/M207G三重変異酵素を設計した9)。ホモロジーモデルを作成して活性中心キャビティの構造を比較してみると、3つのアミノ酸残基の置換により、その大きさが4倍まで拡大することになる (図3)。そこで、本変異酵素を作成したところ、今度はマロニルCoA9分子を縮合して、これまでに例のない非天然型ノナケタイドを生成することを見出した (図2)9)。
一方、本来8分子のマロニルCoAを縮合するOKSについても、2.6 Åの分解能で結晶構造を解明し、PCSのM207G変異酵素とほぼ同一の活性中心キャビティを有することを明らかとした (図3)10)。また、ポリケタイド鎖が伸張していく際に壁となるAsn222残基にGly置換を導入した結果、マロニルCoAの縮合回数が10分子まで拡大して、非天然型ベンゾフェノン誘導体SEK15を単一生成物として高収率で生成することを見出した (図2) 10)。現在、この変異酵素のX線結晶構造解析にも成功し、変異の導入によってキャビティがさらに拡大すること (図3)、さらに、そのポケットの底部を形成するF66残基をN222G変異と同時にLeuに置換することにより、12分子までマロニルCoAの縮合回数が拡大して、非天然型ナフトフェノン誘導体TW95aを生成することも確認している (図2, 3)11)。
5.人工基質をプローブとした非天然型化合物の創出
上述はしなかったが、筆者らは、一方で、化学的に合成した人工基質をプローブとして酵素に作用させることにより、Ⅲ型PKSが異例ともいえる広範な基質特異性と触媒ポテンシャルを示すことを明らかにしてきた2)。例えば、CHSとSTSは、本来、クマロイルCoAを開始基質として、それぞれ、ナリンゲニンカルコンとレスベラトロールへの変換を触媒するⅢ型PKSであるが (図1)、クマロイル基の芳香環の水酸基をフッ素やメトキシ基で置換したエステル、あるいは、ベンゼン環をフランやチオフェンなどヘテロ芳香環で置換した基質アナログを作用させると、全てが酵素反応の開始基質として機能し、マロニルCoAを3分子まで縮合して、一連の非天然型新規化合物を生成する (図4)12,13)。
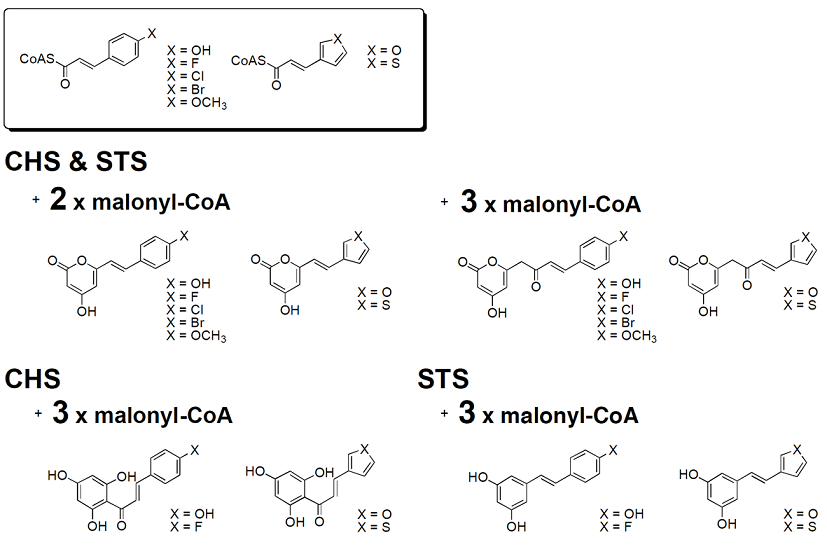

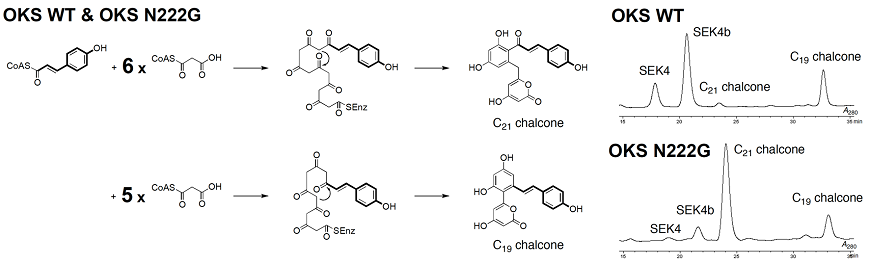
そこで、OKSにクマロイルCoAを開始基質として作用させた結果、クマロイルCoAに5分子あるいは6分子のマロニルCoAが縮合の後、アルドール型縮合により芳香環を形成した、非天然型新規C19スチルベン及びC21カルコンが生成されることを見出した (図5)10)。一方、結晶構造に基づき活性中心キャビティを拡大したOKSのN222G点変異酵素にクマロイルCoAを開始基質として作用させると、生成物の割合が劇的に変化して、C21カルコンの生成量が飛躍的に増大し、主生成物として与えることを見出した (図5)10)。本変異酵素は、C21カルコンの生成により特化したと言える。物質生産を考えるうえで、その生産性の向上も重要な要素であり、点変異の導入により、マロニルCoA縮合回数を増加させただけでなく、基質特異性をも大幅に変化させたことは注目に値する。変異の導入と多様な骨格を有する開始基質や伸長基質を組み合わせることにより、これまで以上に多様性に富んだ化合物ライブラリーの構築が期待される。

6.おわりに
上述したように、Ⅲ型PKSは人為的な酵素機能の制御と分子多様性創出の格好のモデルと言える。また、C, H, O原子で構成される単純な「カルボニルの化学」を触媒するⅢ型PKSに、さらにNなどヘテロ原子を導入した人工基質を作用させれば、N原子の塩基性を利用した新たなC-NあるいはC-C結合の形成も可能になる。実際、すでに筆者らは、N原子を含む人工基質をプローブとすることにより、テトラミン酸骨格や6-5-6縮合環構造のピリドイソインドール骨格、さらには、酵素結晶構造と計算化学を組み合わせ、変異を導入することにより、環拡大した6-7-6縮合環構造をもつ、ジベンゾアゼピン骨格の酵素合成にも成功している。これらについては、追って報告したい。
文献
1) Austin, M. B., Noel, J. P.: Nat. Prod. Rep., 20, 79 (2003).
2) Abe, I., Morita, H.: Nat. Prod. Rep., 27, 809 (2010).
3) Morita, H., Shimokawa, Y., Tanio, M., Kato, R., Noguchi, H., Sugio, S., Kohno, T., Abe, I.: Proc. Natl. Acad. Sci. USA, 107, 669 (2010).
4) Morita, H., Wanibuchi, K., Nii, H., Kato, R., Sugio, S., Abe, I.: Proc. Natl.Acad. Sci. USA, 107, 19778 (2010).
5) Abe, I., Utsumi, Y., Oguro, S., Morita, H., Sano, Y., Noguchi, H.: J. Am. Chem. Soc., 127, 1362 (2005).
6) Abe, I., Oguro, S., Utsumi, Y., Sano, Y., Noguchi, H.: J. Am. Chem. Soc., 127, 12709 (2005).
7) Morita, H., Kondo, S., Oguro, S., Noguchi, H., Sugio, S., Abe, I., Kohno, T.: Chem. Biol., 14, 359 (2007).
8) Austin, M. B., Bowman, M. E., Ferrer, J. L., Schröder, J., Noel, J. P.: Chem. Biol., 11, 1179 (2004).
9) Abe, I., Morita, H., Oguro, S., Noma, H., Wanibuchi, K., Kawahara, N., Goda, Y., Noguchi, H., Kohno, T.: J. Am. Chem. Soc., 129, 5976 (2007).
10) Shi, S.-P., Wanibuchi, K., Morita, H., Endo, K., Noguchi, H., Abe, I.: Org. Lett., 11, 551 (2009).
11) Wanibuchi, K., Morita, H., Noguchi, H., Abe, I.: Bioorg. Med. Chem. Lett., 21, 2083 (2011).
12) Abe, I., Morita, H., Nomura, A., Noguchi, H.: J. Am. Chem. Soc., 122, 11242 (2000).
13) Morita, H., Noguchi, H., Schröder, J., Abe, I.: Eur. J. Biochem., 268, 3759 (2001).
![]()