【トピックス】
二トリルヒドラターゼの反応機構の解明 ―時間分割X 線結晶構造解析による取り組み―
尾高雅文、橋本浩一、養王田正文
東京農工大院・工
1.はじめに
ニトリルヒドラターゼ (Nitrile hydratase; EC4. 2. 1. 84; 以下、NHaseと記す) は1980年に浅野ら (現・富山県立大) によってArthrobacter sp. J-1より発見・命名された酵素1)であり、ニトリルに水を添加してアミドを合成する反応を触媒する (①式)。
R-NC + H2O → R-CONH2 ①
NHaseはRhodococcus、Pseudomonas、Nocardia等に属する多くの微生物から発見・単離されている。NHaseはアミダーゼと同一のオペロン中に存在しており、微生物学的には両酵素によるニトリル化合物の資化 (ニトリルをカルボン酸とアンモニアに分解し、炭素源・窒素源として利用する) を行うと考えられている。また、近年、NHaseの上流の反応を触媒する酵素として、アルドキシムからニトリルを合成する反応を促進するアルドキシム脱水酵素が浅野ら2) と小林 (筑波大) ら3) によって独立に報告され、「アルドキシム-ニトリル経路」の存在が明らかにされ、注目されている。
酵素工学におけるNHaseに関する最も著名な成果はアクリルアミド合成への応用である。京都大学の山田 (現・京大名誉教授) らの研究グループと日東化学工業 (現・三菱レイヨン) らは高いNHase活性をもつ微生物をアクリロニトリルからアクリルアミドを合成する生体触媒として利用することを目指した精力的な研究を行い、1985年にアクリロニトリルからアクリルアミドを合成するバイオリアクターを実用化した。バイオプロセスによる物質生産の多くは医薬品や香料などのファインケミカルであるのに対し、本法は化成品の大量生産に成功した世界初の例として重要である。その後、同グループによって更なる研究開発が進められ、1991年にRhodococcus rhodochrous J1を用いた第3世代のバイオリアクターが実用化され、現在に至っている。アクリルアミドは紙力増強剤や排水処理用の高分子凝集剤、塗料や接着剤などに用いられる重要な化成品であり、年間世界生産量は45万トンを超える。本酵素を用いたアクリルアミド生産量は21世紀に入って飛躍的に増大し、現在では世界生産量の50%以上を占めている。この素晴らしい成果については、既に小林らによる優れた総説4,5) があるので、そちらを参照して欲しい。
筆者らは光応答性鉄型NHaseの反応機構を15年にわたって研究し、最近、反応中間状態の立体構造に基づく触媒反応機構モデルを提唱することに成功した6)。本稿では、NHaseの構造解析に関する研究成果を中心に述べるとともに、最新の触媒反応機構モデルを紹介する。
2.結晶構造解析でNHaseの特異な金属反応中心の構造が明らかに
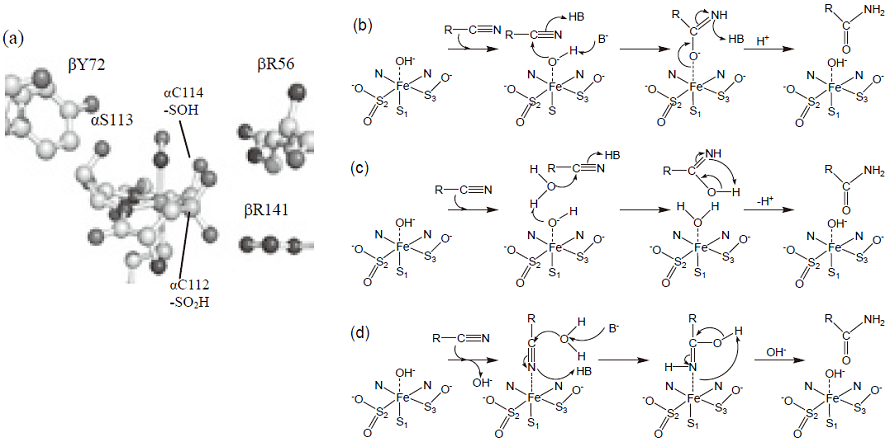
1997年に鉄型NHaseの結晶構造が明らかにされ7)、本酵素はαとβサブユニットのヘテロダイマーが2組会合したヘテロテトラマー構造をとること、金属イオンは両サブユニットの境界面に位置し、αサブユニット上の金属結合モチーフCys1-Xxx-Leu-Cys2-Ser-Cys3に結合していること、配位子はSerとCys3の主鎖アミド窒素、3個のCysのイオウ原子と溶媒の水分子であり、歪んだ6配位構造をとることなどが明らかになった。更に、1998年に筆者を含むグループにより同じ鉄型酵素の高分解能結晶構造が決定され8)、Cys2とCys3がシステインスルフィン酸 (Cys-SO2H) とシステインスルフェン酸 (Cys-SOH) に翻訳後修飾を受けていることが明らかになった (図1(a))。主鎖アミド窒素が金属に直接配位した酵素はNHaseが2例目であり、酸化修飾を受けたシステインを配位子とする酵素はNHaseが始めての報告例である。その後、伏信、若木らのグループによってコバルト型NHaseの結晶構造が決定され9)、更に、一次構造上高い相同性をもつチオシアネート加水分解酵素の結晶構造が筆者らによって明らかにされ10)、鉄型に見られる特異な金属反応中心の構造は、システイン酸化修飾も含めてNHaseと類縁酵素に完全に保存されていることが明らかになった。嫌気的に再構成させた未修飾の鉄型NHaseは触媒活性を示さないが、大気中に放置することで酸化修飾を受け、活性を発現した11)。すなわち、システイン酸化修飾は酵素活性に不可欠であることが判った。
![]()
3.酵素・基質複合体の構造を知る必要 ―時間分割結晶構造解析に向けて―
立体構造が明らかにされると反応機構がわかると思われるかもしれない。しかし、本酵素のように新規構造をもつ場合、むしろ、結晶構造の決定から反応機構研究が本格化する。最初の結晶構造をもとに3つの反応機構が提案された (図1(b)-(d))7)。 (b) と (c) は金属に配位した水分子が直接または近傍の水分子を活性化して基質ポケットに固定されたニトリル炭素原子を求核攻撃するモデルである。これに対し、 (d) では基質のニトリル基が金属イオンに直接配位して活性化され、近傍の水分子による求核攻撃を受ける。いずれの反応モデルにおいても、金属イオンはLewis酸として機能する。しかし、この段階では基質の結合様式すら明らかにされていなかった。
触媒反応機構を理解するためには、酵素・基質複合体の構造を知ることが最も重要である。筆者らはNHaseと基質の複合体結晶を調製し、触媒反応の時間分割結晶構造解析を行うことを考えた。このためには、⑴ 触媒反応を光などの刺激をトリガーとして開始させ、結晶中の各酵素分子の反応を同期させること、⑵ 触媒反応速度を時間分割構造解析の時間分解能に見合う程度に低下させることの2つの条件を満たすことが必要になる。⑴ の条件は鉄型NHaseの光応答性12) を活用できると考えた。筆者らが材料としているRhodococcus erythropolis N771由来鉄型NHase (ReNHase) は、活性中心である非ヘム鉄に一酸化窒素 (NO) を結合させる (ニトロシル化) ことによって不活性化し、NOを光解離させることで瞬時に再活性化できる13)。問題は ⑵ の条件であった。現在、X線結晶構造解析で1データセットの測定に要する最短時間はおよそ10分程度である。これに対してニトリル水和反応のkcatは約1000 s-1であり、106倍近く早い。そのため、反応速度を著しく低下させた実験系が必要であった。
4.反応速度が遅い基質の探索
これまでに報告されたNHaseの基質選択性を調べたところ、イソブチロニトリル (iBuCN) とイソバレロニトリル (iVCN) は極めて反応性が乏しく、Ki=5µM程度の競争阻害活性を示すことが判った。そこで、これら2つのニトリルを候補とし、研究を開始した。
まず、iBuCNとReNHaseとの相互作用を詳細に解析した。その結果、予想外にiBuCN自体はノーマルな基質であり、市販試薬中に混入している不純物が高い阻害活性を示すことが判った。この不純物を単離同定したところ、2-cyano-2-propyl hydroperoxide (Cpx) という、iBuCNの2位の水素がヒドロペルオキシド基に置換された化合物が阻害物質であることが明らかになった14)。Cpxは化学量論比1:1の添加でReNHaseのCys-SOH配位子 (αCys114-SOH) をCys-SO2Hに特異的に酸化し、失活させた。すなわち、αCys114がCys-SOHの酸化状態をとることが酵素活性の発現に重要であることが明らかにされた。
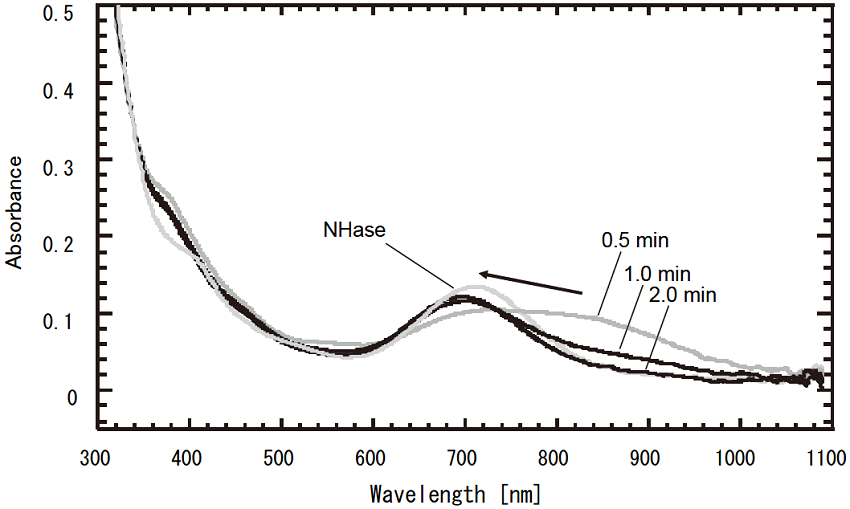
市販iVCNをReNHaseに添加すると、非ヘム鉄センターに特徴的な680 nmの吸収スペクトルが820 nm付近にシフトし、数分後に回復した (図2)。この結果は市販iVCNまたは試薬に含まれる不純物が非常にゆっくりと反応する可能性を示している。詳細な解析の結果、市販試薬に混入しているイソブチロイソニトリル (CH3CH(CH3)CH2NC; iBuNC) が遅い反応をする正体であることが判った。iBuNCの反応生成物を質量分析法によって解析したところ、イソブチルアミン (CH3CH(CH3)CH2NH2; iBuNH2) であった。同様な反応は他のイソニトリルでも観測され、ReNHaseはイソニトリルをアミンに加水分解する新規な酵素活性をもつことが判った15)。イソニトリルは極めて高い競争阻害活性を示すことから、ニトリルと同様の触媒反応過程を経る可能性が高い。そこで、イソニトリルを基質としてReNHaseの時間分割構造解析を行うことを考えた。iBuNCは化学的に不安定なため、以降の反応はtert-ブチルイソニトリル (tBuNC) を用いることにした。イソニトリル炭素由来の生産物をATR-FTIRで分析したところ、一酸化炭素 (CO) であることが判った。すなわち、ReNHaseは②式に示す新規反応を触媒する。
tBuNC + H2O → tBuNH2 + CO ②
![]()
キネティクス測定から、tBuNC加水分解反応のKmはニトリル水和反応と同程度であるが、kcatは1.8×105倍小さい値であった。以上より、tBuNCは時間分割構造解析に適した基質であると期待された6,15)。
5.tBuNC加水分解反応の時間分割結晶構造解析6)
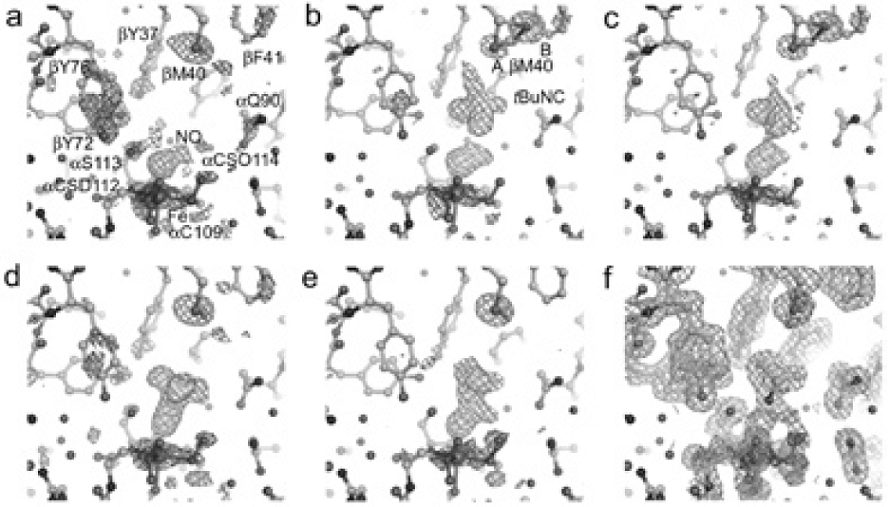
NO結合型ReNHase結晶に暗所でtBuNCを蒸気拡散によって浸漬させ、反応開始前の結晶とした。20℃で結晶に冷光照明を照射して開始させた。一定時間後、低温の窒素ガスを吹き付けて反応を停止させ、結晶構造を決定した。全ての構造で基質ポケット以外に大きな構造変化は無く、αCys-SO2H (αCSD112) とαCys-SOH (αCSO114) の翻訳後修飾はすべての構造において確認された (図3)。光活性化440分以降では、結晶へのダメージが蓄積してしまい、構造を決定できなかった。
⑴ 光照射前 (反応開始前)
基質を添加する前の構造は既に決定されているニトロシル型酵素の結晶構造と極めて良く似ていた (図3(a))。tBuNCを加えた結晶では、tBuNC分子の電子密度が基質ポケット内に明瞭に確認され、–NC基はFe3+に対して逆側に配置していた (図3(b))。疎水性の高い基質ポケットにtBuNCの3級ブチル基が入り、かつ、反応中心のFe3+にはNOが配位しているため、-NC基が空間的に余裕のある位置で安定化したと考えられる。
⑵ 光照射開始から120分後まで
光照射開始18分後では、NOとtBuNCに対応する電子密度図、特にイソニトリル基に対応する電子密度図が減少した (図3(c))。120分後では、NOに相当する電子密度が消失し、代わりにtBuNC分子がFe3+とFe-C(-NC)=2.06Åの距離で結合していた (図3(d))。基質のイソニトリル基はFe3+に直接配位することがわかった。
⑶ 光照射120分後から440分後まで
440分 (図3(e)) におけるイソニトリル分子由来の電子密度図は120分後のものとは明確に異なっていた。3級ブチル基に対応する電子密度図は120分のときのFe3+の位置から約1.0Å離れており、イソニトリル基の炭素原子とαCys114-SOHのOδ原子に対応する電子密度図との間に溶媒の水分子由来と思われる新たな電子密度図が観測された。すなわち、この時間に触媒反応が進行していることが予測される。そこで、tBuNH2の存在を仮定して440分の電子密度図を再計算させたところ、得られた構造は電子密度図によく一致した (図3(f))。

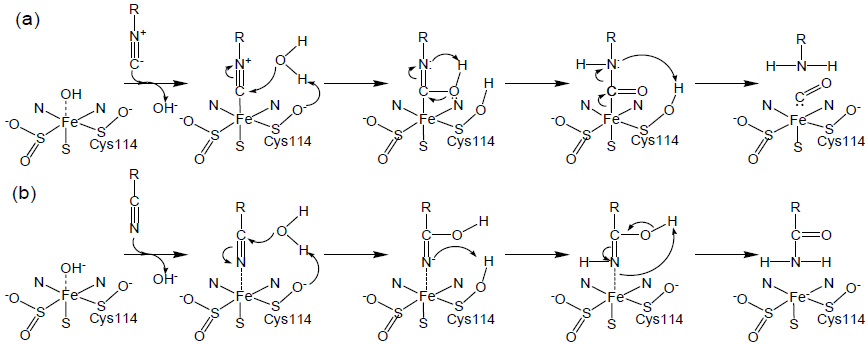
6. 時間分割X線結晶構造解析の結果から予想される触媒反応機構
以上の結果から予想されるtBuNC加水分解の触媒反応機構を図4(a) に示す。tBuNCは金属に配位した後、αCys114-SOHのOδ原子によって活性化された水分子の求核攻撃を受け、tBuNH2とCOを生じる。イソニトリルとニトリルとの構造の類似点を考慮すると、ニトリルの水和反応も同様に進行すると予想される (図4(b))。すなわち、ニトリル水和反応は前述した図1(d) の反応モデルに従い、Fe3+はニトリル基の窒素原子を配位してニトリル炭素を活性化するとともに、ニトリル基をFe3+上方のキャビティに固定する。その後、αCys114-SOHのOδ原子によって活性化された水分子がニトリル基の炭素原子を求核攻撃することで水和反応が進行する。イソニトリル加水分解反応のkcatが著しく小さい理由は、αCys114-SOHのOδ原子が立体障害となるために、H2Oaがイソニトリル基の炭素原子に近づきにくいためではないかと思われる。NHase触媒反応におけるCys-SOH基の重要性はモデル錯体による研究からも報告されている。Na[Co(L-N2SOSO)(tBuNC)2]と(Me4N)[Co(LN2SO2SO2)(tBuNC)2]というイオウの酸化状態のみが異なるモデル錯体では、前者のみがニトリル水和活性を示した16)。最近、名古屋工大の増田らはイオウ配位子の酸化状態が異なるN2S2(tBuNC)2型Co錯体を合成・解析し、システイン配位子の酸化に伴ってCo3+のLewis酸性が増大すること、SO配位子のみが溶媒に対して求核的性質を示すことを報告した17)。また、最近報告された理論計算による研究においても、基質の鉄イオンへの配位を仮定した場合、αCys114-SOHがCatalytic Baseとして機能し、溶媒の水分子からプロトンを引き抜く可能性が指摘されている18)。一方、近年、Holzらはニトリルヒドラターゼの触媒活性の温度またはpH依存性を詳細に解析し、金属配位子のセリンと周辺に存在するチロシンやトリプトファン残基がCatalytic Triardを形成することを主張している19,20)。実際、結晶構造からCatalytic Baseとして予測されるのは、このセリン残基のヒドロキシル基、近傍にあるチロシン残基のフェノール性水酸基とαCys114-SOHのスルフェニル基のみである。筆者らの研究室において、このセリンとチロシン残基をそれぞれアラニンとフェニルアラニンに置換した変異体を作製したところ、ともに立体構造にはほとんど影響しないが、NOとの親和性が大きく低下し、また、触媒活性も大きく損なわれることがわかっている (山中ら、投稿中)。したがって、提唱されているNHaseの触媒反応モデルを検証するためには、今後、反応速度を低下させた変異体を用いてニトリルを基質とした時間分割構造解析を行うとともに、得られた反応機構を量子化学計算等で解析して行くことが必要である。また、構造では明らかにできない金属反応中心の電子状態を振動分光やX線吸収スペクトル等で解析してゆくことも重要である。さらに、この反応モデルではαCys114-SOHが脱プロトン化し、αCys114-SO-となっていることが重要である。しかし、スルフェニル基の脱プロトン化に関しては、これまでのところ相反する結果が報告されている。これらの問題に決着を付けるためには、中性子構造解析等、最新の構造解析手法を行う必要があると考えている。冒頭で触れたように、本酵素は我が国の研究者の手で発見・命名され、極めて有用な工業利用が実現され、新規な翻訳後修飾を含む反応中心の構造が明らかにされた我が国オリジナルとも言えるタンパク質である。今回、有力な反応モデルを提唱したことで、触媒反応機構の研究に関しても他国を大きくリードすることができたと考えている。さらに、最近、本酵素のサブユニットが集合する際に金属結合サブユニットの交換が起きる (Self-Subunit-Swapping) ことが小林らによって発見され21,22)、金属酵素の成熟機構に関しても、我が国から新たな成果が報告されている。本酵素の主要な研究成果が全て日本発となる日が来ることを強く期待している。
![]()
謝辞
本稿で述べた研究は理化学研究所に所属していたときに、遠藤勲主任研究員のご指導のもと、長棟輝行博士 (現・東大)、神谷信夫博士 (現・大阪市大) らと共同で開始した研究を発展させたものである。時間分割X線結晶構造解析の研究は神谷信夫博士、河野能顕博士 (理研・播磨研)、野尻正樹博士 (現・阪大) らの先行研究によるところが大きい。また、本稿で述べた新規酵素活性の発見は谷口佳代子博士 (現・東京農大) によるものであり、その解析では、野口 巧博士、鈴木博行博士 (筑波大)、中村健道博士 (理研)、高橋俊哉博士 (理研) に大変お世話になった。上記を始めとする共同研究者の皆様へ深く感謝、御礼申し上げます。
文献
1) Asano, Y., Tani, Y., Yamada, H.: Agric. Biol. Chem., 44, 2251 (1980).
2) Asano, Y., Kato, Y.: FEMS Microbiol. Lett., 158, 185 (1998).
3) Xie, S.-X., Kato, Y., Komeda, H., Yoshida, S., Asano, Y.: Biochemistry, 42, 12056 (2003).
4) Kobayashi, M., Nagasawa, T., Yamada, H.: Trends Biotechnol., 10, 402 (1992).
5) Kobayashi, M., Shimizu, S.: Nature Biotechnol., 16, 733 (1998).
6) Hashimoto, K., Suzuki, H., Taniguchi, K., Noguchi, T., Yohda, M., Odaka, M.: J. Biol. Chem., 283, 36617 (2008).
7) Huang, W., Jia, J., Cummings, J., Nelson, M., Schneider, G., Lindqvist, Y.: Structure, 5, 691 (1997).
8) Nagashima, S., Nakasako, M., Dohmae, N., Tsujimura, M., Takio, K., Odaka, M., Yohda, M., Kamiya, N., Endo, I.: Nat. Struct. Biol., 5, 347 (1998).
9) Miyanaga, A., Fushinobu, S., Ito, K., Wakagi, T.: Biochem. Biophys. Res. Commun., 288, 1169 (2001).
10) Arakawa, T., Kawano, Y., Kataoka, S., Katayama, Y., Kamiya, N., Yohda, M., Odaka, M.: J Mol Biol., 366, 1497 (2007).
11) Murakami, T., Nojiri, M., Nakayama, H., Odaka, M., Yohda, M., Dohmae, N., Takio, K., Nagamune, T., Endo, I.: Protein Sci., 9, 1024 (2000).
12) Nagamune, T., Kurata, H., Hirata, M., Honda, J., Koike, H., Ikeuchi, M., Inoue, Y., Hirata, A., Endo, I.: Biochem. Biophys. Res. Commun., 168, 437 (1990).
13) Odaka, M., Fujii, K., Hoshino, M., Noguchi, T., Tsujimura, M., Nagashima, S., Yohda, M., Nagamune, T., Inoue, Y., Endo, I.: J. Am. Chem. Soc., 119, 3785 (1997).
14) Tsujimura, M., Odaka, M., Nakayama, H., Dohmae, N., Koshino, H., Asami, T., Takio, K. Yoshida, S., Maeda, M., Endo, I.: J. Am. Chem. Soc., 125, 11532 (2003).
15) Taniguchi, K., Murata, K., Murakami, Y., Takahashi, S., Nakamura, T., Hashimoto, K., Koshino, K., Dohmae, N., Yohda, M., Hirose, T., Maeda, M., Odaka, M.: J. Bioeng. Biosci., 106, 174 (2008).
16) Heinrich, L., Mary-Verla, A., Li, Y., Vassermann, J., Chottard, J. C.: Eur. J. Inorg. Chem., 9, 2203 (2001).
17) Yano, T., Wasada-Tsutsui, Y., Arii, H., Yamaguchi, S., Funahashi, Y., Ozawa, T., Masuda, H.: Inorg. Chem., 46, 10345 (2007).
18) Hopmann, K. H., Guo, J. D., Himo, F.: Inorg. Chem., 46, 4850 (2007).
19) Mitra, S., Holz, R. C.: J. Biol. Chem., 282, 7397 (2007).
20) Rao, S., Holz, R. C.: Biochemistry, 47, 12057 (2008).
21) Zhou, Z., Hashimoto, Y., Shiraki, K., Kobayashi, M.: Proc. Natl. Acad. Sci. USA, 105, 14849 (2008).
22) Zhou, Z., Hashimoto, Y., Kobayashi, M.: J. Biol. Chem., 284, 14930 (2009).
![]()