【トピックス】
タンパク質ジスルフィド結合の形成に関わる酵素群
― 構造が明らかにしたそのメカニズム―
稲葉謙次
九大・生体防御医学研
1.はじめに
今から40年以上も前に、Anfinsenらによる還元型リボヌクレアーゼを用いた変性再生実験によって、ジスルフィド結合の形成を伴う蛋白質の酸化的フォールディング反応は、適度な酸化還元条件下では、自発的に進行することが示された1)。しかし細胞の中では、これがより迅速かつ正確に進行するよう、幾つかのジスルフィド結合導入酵素・異性化酵素が存在することが明らかとなってきた。特に1990年代に入り、大腸菌や出芽酵母などのこれに関わる因子が次々と発見され、基質から最終電子受容体までの電子カスケードの全体像が記載されるに至っている2,3)。
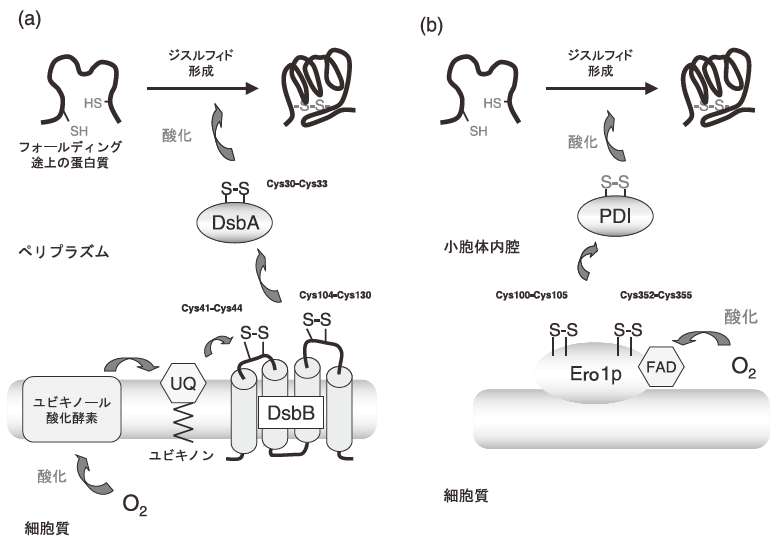
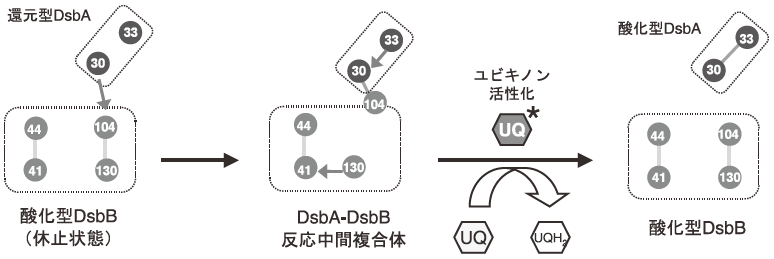
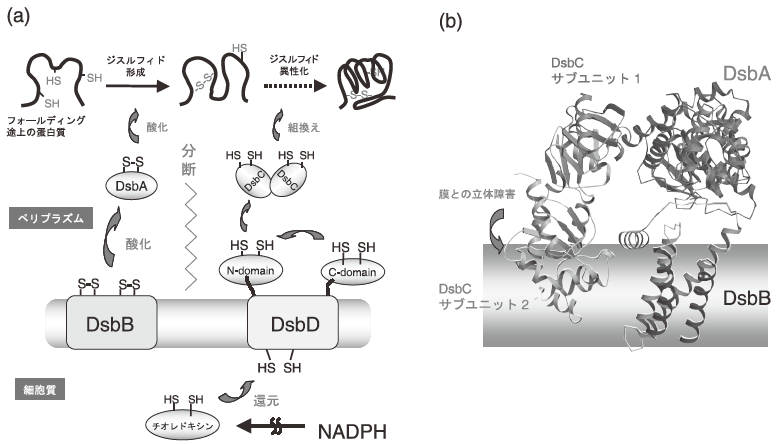
グラム陰性細菌である大腸菌では、主にペリプラズム (内膜と外膜の間のスペース) において蛋白質ジスルフィド結合は形成される。還元的な環境にある細胞質では通常ジスルフィド形成反応は進行しないが、酸化ストレスなどに応答して一部の転写因子・分子シャペロンが一過的にジスルフィド結合を形成する例は知られている。ペリプラズムにはジスルフィド結合導入酵素としてDsbAが存在し、基質を酸化し還元型となったDsbA は内膜蛋白質DsbB により再酸化される。DsbBが受け取った電子は呼吸鎖成分であるユビキノン (UQ) に受け渡され、最終的にはユビキノール酸化酵素を介して酸素が電子受容体となる (図1a)4,5)。ペリプラズムをもたないグラム陽性細菌においても、大腸菌のDsbA、DsbBに機能的に対応する酸化酵素が細胞表層に存在することがすでに知られている。
一方、出芽酵母などの真核細胞では、酸化的な蛋白質のフォールディングは主に小胞体内腔において進行する。そこにはジスルフィド結合導入酵素としてProtein Disulfide Isomerase (PDI) が存在し、基質を酸化し還元型となったPDIは膜表在性のEro1pにより再酸化される (図1b)。Ero1pはFAD結合蛋白質であり、おそらくFADは電子受容体として機能している。このように、大腸菌におけるDsbA-DsbB-UQ酸化システムと真核細胞のPDI-Ero1p-FADは、互いに機能的ホモログの関係にある。何れの場合も、ジスルフィド結合の形成は、エネルギー代謝のキーとなる分子に依存した細胞の酸化還元系との共役により行われている。
![]()
2.大腸菌におけるジスルフィド結合導入酵素:DsbA
DsbAは、フォールディング途上の基質タンパク質のシステインペアーを酸化する (つまりジスルフィド結合を導入する) 可溶性ペリプラズム酵素である。dsbA遺伝子の機能欠損変異株では様々な分泌タンパク質がジスルフィド結合を欠く状態となる。DsbAはチオレドキシンフォールドを有し、その中に酸化還元活性を担うCys30-Pro-His-Cys33配列を持つ (図2)。Cys30は低いpKa値 (~3.5) を持ち、反応性が高い。DsbAの酸化還元電位 (-0.089 V) は、知られているジスルフィド酸化還元酵素の中で最も高い (酸化力が強い)。酸化還元電位的には還元型を好むDsbAは、細胞内でDsbBの働きにより酸化型に保たれることによってジスルフィド結合導入酵素としての機能を発揮できる。DsbAの結晶構造によれば6)、その活性部位 (Cys30-Cys33ペア) の近傍には、基質ポリペプチドの結合部位と考えられる疎水性の溝が存在する (図2)。DsbAが基質のシステインペアを酸化するとき、まず、Cys30と基質システインの一つとの間に分子間ジスルフィド中間体を作る。DsbAのCys30-Cys33ペアは酸化還元電位が高い (還元型になりやすい) ため、この中間体は基質側のもう一つのシステインからの求核攻撃をうけて開裂し、ジスルフィドがDsbAから基質の方に移る (図2下)。
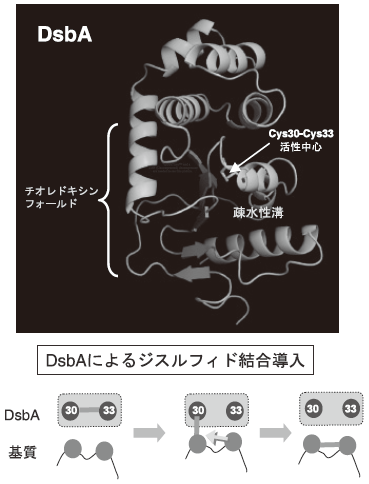
図2 DsbAの結晶構造6)とジスルフィド結合導入の機構
3.DsbA酸化膜酵素:DsbB
著しく酸化力の強い (酸化しがたい) DsbAを酸化状態に保つ機構が、ジスルフィド結合導入システムにおける最も本質的な問題の一つである。細胞質膜の内在性膜蛋白質であるDsbBがその役割を果たす。DsbBのペリプラズムに露出した領域にはCys41-Cys44及びCys104-Cys130の二組のシステインペアが存在し,休止状態では共にジスルフィド結合を形成している (図1a)。興味深いことに、DsbBのこれら二つのシステインペアは共に、酸化還元低分子化合物との化学平衡により計測する限り、DsbAのCys30-Cys33ペアよりも低い酸化還元電位をもつ7,8)。通常電子は酸化還元電位の低いところから高いところへ移動するため、DsbAからDsbBへの電子移動反応は単純な酸化還元電位の観点からは説明しがたい (6節参照)。
DsbBのもう一つの特徴は、酸化力の源であるユビキノンと結合していることである。ユビキノンはDsbBのCys41-Cys44ペアを特異的に酸化することで、キノンの酸化力が蛋白質ジスルフィド結合という形に変換される。ここで著者らは、DsbB上でジスルフィド結合が最初に創りだされる過程において、500 nm 付近に強い吸収極大を示すCys44-UQ間の電荷移動錯体が過渡的に形成されることを発見している9)。そして量子化学シミュレーションの結果と統合することにより、図3に示すジスルフィド結合創生のための化学スキームを提唱している10)。
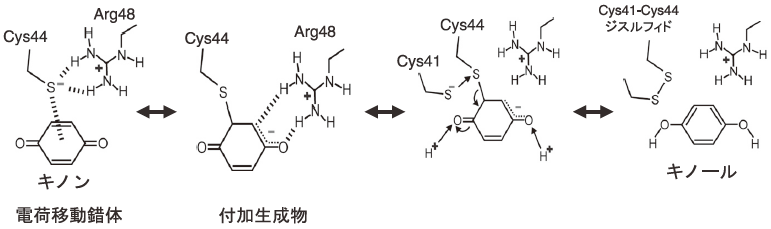
図3 DsbBとキノンによるジスルフィド結合新規創生の化学スキーム10)
4.DsbA-DsbB-UQ複合体の結晶構造
以上記述したDsbA-DsbB-UQから構成される酸化システムの機能発現メカニズムを深く理解する上で、その三次元構造は何よりも必要不可欠な情報である。しかしながら、DsbBは膜蛋白質であり、その結晶構造解析は著者らのグループに限らず海外のグループでも長年困難を極めていた。そこで著者らは、あえて水溶性酵素であるDsbAとの複合体の状態でDsbBの結晶構造解析を目指すことにした。成功に至るまでの詳細については原著論文を参照していただくとして、約4年間の努力が実り、DsbA-DsbB-UQ複合体の結晶構造解析に成功した11)。アミノ酸配列から予測された通り、DsbBは4本の膜貫通へリックス (TM1~TM4) から成り、Four-Helix Bundleの基本骨格をもつ。さらにDsbBは、TM3からTM4にかけての長いペリプラズム側ループの中に、意味ありげに膜に水平方向のαヘリックスを一本もつ (図4a)。この膜水平ヘリックスからTM4にかけての領域は電子密度が欠失しており、この領域は非常に動きやすい性質をもつと考えられる。電子受容体として機能する補酵素UQはDsbBのFour-Helix Bundleの中、TM2のN末端近傍に位置する (詳細は次節参照)。
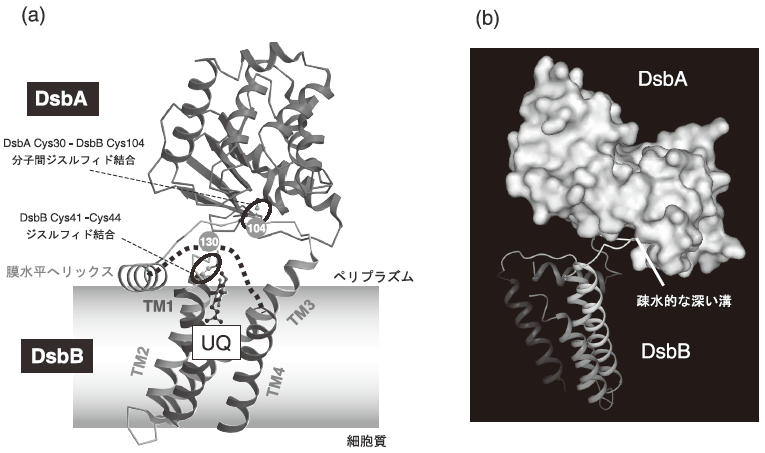

一方DsbA領域については、DsbBとの複合体のときと単独のときとで、構造上大きな違いはなかった。注目すべきことに、DsbAにはフォールディング途上の基質と結合すると考えられている疎水的な深い溝が存在するが、DsbBのCys104を含む伸びたセグメントは、まさにこの溝に引っ張られる形でDsbAと結合している (図4b)。おそらくDsbAは、DsbBのこの高次構造に欠けた領域を、フォールディング途上の基質蛋白質かのごとく認識しているのであろう。
5.UQ結合部位
この構造解析における最も重要な発見の一つが、DsbB上のUQ結合部位である。TM2のN末端近傍にUQのリング構造にフィットした電子密度が観測されたが (図5)、3.7 Åという分解能ではこれをUQと断定することはできない。そこで著者らはUQ非結合型の結晶も作製し、UQ結合型の結晶との差フーリエマップを算出した。その結果、UQ依存的に差の電子密度が予測した位置に現れ、UQの結合部位を特定することができた。著者らが同定したUQの近傍にはCys41-Cys44ペアが存在し、このことはUQがCys41-Cys44ペアを特異的に酸化するという過去の実験結果とも合致する12)。さらに、この構造情報は図3に示す化学スキームが実際にDsbB上で起こっていることを強くサポートする。実際、Cys44はUQと電荷移動錯体を形成できる所に位置する。またDsbBホモログ間で高度に保存され、ジスルフィド結合創生反応おいて触媒残基として必須であることが判明しているArg48の側鎖10) も図3のスキームと合致した場所に存在する。まさに生化学実験・量子化学シミュレーションの統合により提唱されたDsbB上でのジスルフィド結合創生のメカニズムが、構造的側面から証明されたのである。Cys41, Cys44, Arg48そしてUQにより構成されたこの活性中心は、細胞の中で次から次へとジスルフィド結合を産生する反応場と捉えることが出来る。
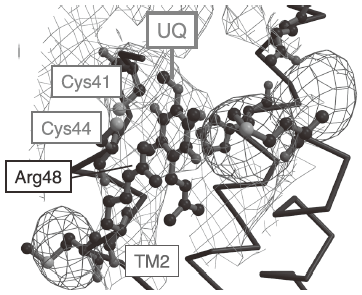
図5 DsbB上のユビキノン (UQ) 結合部位
6.DsbBによるDsbA酸化の分子機構
3節でも触れたように、DsbA-DsbB-UQ酸化システムの最も難解かつ興味深い問題は、相手を酸化する能力に優れた (つまり自身は酸化を受けにくい) ジスルフィド結合をもつDsbAが、DsbBにより如何に効率よく酸化されるかにある。実際、DsbBの各システインペア (Cys41-Cys44およびCys104-Cys130) はDsbAより弱い酸化能力しかもたないにもかかわらず、DsbBはユビキノン非依存的にDsbAを酸化する能力をもつことを確認している8)。
この残された重要問題についても、今回の構造解析はもっともらしい解答を与えてくれる。DsbBはDsbAと出会う前、通常Cys41-Cys44ペアおよびCys104-Cys130ペアの間で二組のジスルフィド結合を形成している。ところが複合体の構造を眺めると、Cys104とCys130はもはやジスルフィド結合を形成できない距離にまで互いに離れ、しかもフリーになったCys130はCys41-Cys44ジスルフィドに近接している。ちなみに電子密度が欠けた領域に含まれるCys130の位置は、ここにSeMetを導入しSeの異常分散ピークを観測することにより決定した。Cys104-Cys130間が距離的に離れることは、図4bで示されるように、DsbAがDsbBのCys104を含む数残基のペプチドを自身の深い溝に引っ張り込むことに起因すると考えている。またDsbBのCys104とCys130の間にはDsbA Met64の側鎖が介在していることも確認しており、その寄与も見逃せない。
では何故、複合体形成によりCys104とCys130が十分に離れることが、DsbBによるDsbA酸化反応において好都合なのであろうか。図6に示すように、この反応は還元型DsbAの反応性に富んだCys30がDsbBのCys104-Cys130ジスルフィドを特異的に求核攻撃し、分子間ジスルフィド結合を形成することから始まる。肝心なのは次のステップである。本来酸化還元電位の性質を考慮すれば、DsbBよりもDsbAの方が還元型で存在することを好むので、上記の中間複合体が生じたとしても、また逆反応により最初の状態に戻る傾向にあると考えられる。ところが逆反応が起こるためには、フリーになったCys130が分子間ジスルフィド結合を求核攻撃しなければならない。Cys104とCys130が隔離されていることは、まさにこの逆求核攻撃を抑制する上で重要な意味をもつと考えられる。さらに、Cys130はもう片側のCys41-Cys44ジスルフィドに近接しており、このことは実際に生体内で起こるCys104-Cys130ペアからCys41-Cys44ペアへの電子移動反応を促進する上で好都合である。実際、一連のDsbA酸化反応において、DsbB内でCys41-Cys130間のジスルフィド結合が過渡的に形成することは実験的に示されている9,13)。以上のように、DsbBは特別な基質であるDsbAと出会うことにより巧妙にCysの配置を変化させ、DsbAだけを選択的に酸化するように設計されているのである。言い換えれば、DsbBはDsbAに特化した形で構造変化を引き起こし、超強力酸化酵素に変身するのである。

7.DsbA-DsbB 酸化経路とDsbC-DsbD還元経路の分断機構
我々が解いた複合体の構造は、さらにもう一つ生物学的に重要な知見を含む。DsbAはジスルフィド結合導入酵素としては優れているが、導入すべきシステインペアを選択する能力には欠ける。そこで大腸菌のペリプラズムには、誤ったジスルフィド結合を組換える酵素DsbCが存在する。DsbCが異性化酵素として機能するためには、活性部位は還元型に保たれていなければならないが、その還元力の源は細胞質に存在するチオレドキシンに由来している。チオレドキシンからの還元力は内膜タンパク質DsbDを介してDsbCへ受け渡される (図7a)。DsbCもDsbAと類似のチオレドキシンドメインを有しているが、DsbAはモノマーで機能しているのに対し、DsbCはV字型のホモダイマーを形成している。ここで極めて重要なことは、ジスルフィド結合導入のためのDsbA-DsbB酸化経路とジスルフィド結合組換えのためのDsbC-DsbD還元経路がクロストークしないよう、分断されていることである。なぜなら、仮にDsbBがDsbCを速やかに酸化すれば、DsbCはもはや異性化酵素として働けないからである。
![]()
そこで、今回解いたDsbA-DsbB複合体の構造上でDsbAおよびDsbCのチオレドキシンドメインを重ね合わせると、DsbCのもう片側のサブユニットが膜に衝突することが予想された (図7b)。この立体障害により、DsbCはDsbBにアクセスできず、両経路が分断されていると解釈できる。実際、モノマー構造をとるようになったDsbCの変異体はDsbAに代わってDsbB依存的なジスルフィド導入酵素として機能し得ることが他のグループにより報告されており14)、このことは上で述べた分断機構を実験的に強くサポートする。
8.真核細胞のジスルフィド結合創生システムとの類似点
“はじめに”でも触れたように、真核細胞の小胞体におけるジスルフィド形成はUQではなくFADに依存している。つまり、DsbBの機能ホモログであるEro1pはFADに依存してジスルフィド結合を創り出す。膜内在性のDsbBは脂溶性のUQを補酵素として用いているのに対し、活性ドメインを膜外にもつEro1pが水溶性のFADを補酵素としていることは、理にかなっている。Ero1pの膜外ドメインの結晶構造はDsbBよりも前に2004年に報告されている15)。興味深いことに、Ero1pも活性部位近傍はFour-helix Bundleの基本骨格をもち、FADはその中央部に結合している (図8)。FADの近傍にはCys352-Cys355ペアが存在し、このシステインペアがFAD依存的にジスルフィド結合を形成すると考えられる。一方PDIと直接相互作用するのはCys100-Cys105ペアと考えられており、このペアは非常にフレキシブルなループ領域に存在する。このように水溶性ドメイン、脂溶性のドメインの違いはあるものの、Ero1pの基本骨格およびその中に存在する補酵素およびシステインペアの配置は、DsbBのそれとほぼ同一である16)。ちなみに、DsbBとEro1pの間にはアミノ酸配列の相同性がほとんどない。
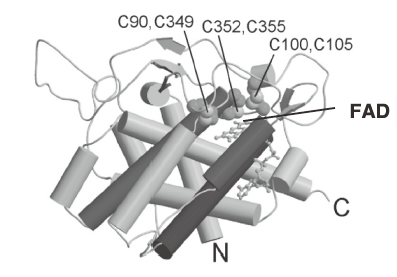
図8 Ero1pの膜外ドメインの結晶構造15)
さらに興味深いことに、Ero1pがモデル基質であるチオレドキシンと酸化還元反応を行うと、Ero1pに結合したFADの吸収極大が長波長シフトすることが観測されている17)。まさにDsbBで観測されたUQの電子状態遷移と同様の現象である。このように、大腸菌のDsbB-UQシステムと真核細胞のEro1p-FADシステムは、役者は違えど、全体構造から化学スキームに至るまで多くの共通点がある。真核細胞と原核細胞の間でそれぞれ独立してジスルフィド結合創生システムが進化したとすれば、その根本となる原理を共有していることは、上記の基本骨格および反応機構の生物学的および化学的合理性を暗示している。
9.おわりに
以上解説したように、大腸菌および出芽酵母において蛋白質ジスルフィド結合の形成に関わる細胞因子の構造解析が目覚ましく進展し、分子機構の詳細さえも多く語れるようになった。これら細胞が有するジスルフィド結合形成補助システムを深く理解することは、複数のジスルフィド結合を有する有用な酵素・蛋白質性ホルモン・抗体などの高効率再構成という工学的利用につながる。一方高等生物細胞においては、まだまだ多くの酸化還元関連因子が機能未知のまま残されている。チオール・ジスルフィドの交換を介したネットワークは細胞における蛋白質の品質管理と密接に関連している。したがって、このネットワークの機能発現機構の解明は、ミスフォールド蛋白質や過度の酸化ストレスが引き起こす様々な疾患の治療にも将来的にはつながり、その医学的貢献は計り知れない。今後も、分子生物学・生化学・構造生物学などの多角的アプローチにより開拓すべき重要研究課題の一つであると確信している。
謝辞
ここで紹介した研究は全て京都大学ウイルス研究所伊藤維昭教授の研究室で遂行したものである。DsbA-DsbB複合体のX線結晶構造解析は、村上聡博士 (阪大・産研)、鈴木守博士、中川敦史博士、山下栄樹博士、月原冨武博士 (以上、阪大・蛋白研)、岡田健吾博士 (奈良先端大) との共同で行ったものである。量子化学シミュレーションについては、林重彦博士 (京大・理) との共同で行ったものである。これら全ての研究者に、この場を借りて厚く深く御礼申し上げます。
文献
1) Anfinsen, C. B., Haber, E., Sela, M., White, F. H.: Proc. Natl. Acad. Sci. USA, 47, 1309 (1961).
2) Kadokura, H., Katzen, F., Beckwith, J.: Ann. Rev. Biochem., 72, 111 (2003).
3) Sevier, C. S., Kaiser, C. A.: Nat. Rev. Mol. Cell. Biol., 3, 874 (2002).
4) Kobayashi, T., Kishigami, S., Sone, M., Inokuchi, H., Mogi, T., Ito, K.: Proc. Natl. Acad. Sci. USA, 94, 11857 (1997).
5) Bader, M., Muse, W., Ballou, D. P., Gassner, C., Bardwell, J. C. A.: Cell, 98, 217 (1999).
6) Martin, J. L., Bardwell, J. C. A., Kuriyan, J.: Nature, 365, 464 (1993).
7) Inaba, K., Ito, K.: EMBO J., 21, 2646 (2002).
8) Inaba, K., Takahashi, Y.-h., Ito, K.: J. Biol. Chem., 280, 33035 (2005) .
9) Inaba, K., Takahashi, Y.-h., Fujieda, N., Kano, K., Miyoshi, H., Ito, K.: J. Biol. Chem., 279, 6761 (2004).
10) Inaba, K., Takahashi, Y.-h., Ito, K., Hayashi, S.: Proc. Natl. Acad. Sci. USA, 103, 287 (2006).
11) Inaba, K., Murakami, S., Suzuki, M., Nakagawa, A., Yamashita, E., Okada, K., Ito, K.: Cell, 127, 789 (2006).
12) Kobayashi, T., Ito, K.: EMBO J., 18, 1192 (1999).
13) Kadokura, H., Beckwith, J.: EMBO J., 21, 2354 (2002).
14) Bader, M. W., Hiniker, A., Regeimbal, J., Goldstone, D., Haebel, P. W., Riemer, J., Metcalf, P., Bardwell, J. C. A.: EMBO J., 20, 1555 (2001).
15) Gross, E., Kastner, D. B., Kaiser, C. A., Fass, D. A.: Cell, 117, 601 (2004).
16) Sevier, C. S., Kadokura, H., Tam, V. C., Beckwith, J., Fass, D., Kaiser, C. A.: Protein Science, 14, 1630 (2005).
17) Gross, E., Sevier, C. S., Heldman, N., Vitu, E., Bentzur, M., Kaiser, C. A., Thorpe, C., Fass, D.: Proc. Natl. Acad. Sci. USA, 103, 299 (2006).
![]()