【トピックス】
生体触媒を用いたα-置換カルボン酸のデラセミ化反応
―ラセミ体から光学活性体への高効率変換法―
加藤太一郎
兵庫県大院・工
1.はじめに
鏡像異性体同士を分離することは容易ではない。それは沸点や溶解度といった物理的性質や化学的性質は同じであり、違っているのは光学的性質、つまり旋光度の符号だけだからである。しかし作用する相手が光学活性体になると話は変わってくる。つまり光学活性体を作用させた場合の応答は異性体間で同じとは限らない。これは光学異性体の重要な特徴のひとつである。
タンパク質は光学活性なアミノ酸から構成されており、キラルな環境場を形成している。そのため光学活性化合物の両鏡像体を厳密に区別する場合が多い。例えばインスタントのまつたけのお吸い物に添加されているマツタケオール (1-octene-3-ol) という化合物は、S体はまつたけの香りを有するがR体は草のにおいがする。もしこの添加物が2つの鏡像体の混合物で作られたなら、それは商品として成り立たなくなってしまう。
また、世界の流れは環境低負荷型社会の形成に進んでおり、不要なものをできるだけ使わない、作らないということが重要視されつつある。どのような方法で作るかということがこれまで以上に重要になってきている。既存の化学的手法による物質生産に代わってバイオプロセスを導入する企業が多いのもそのような理由からであろう。本稿では生体触媒を用いて光学活性体を効率よく調製する手法を紹介する。本法はデラセミ化反応と呼ばれ、ラセミ体を出発物質として最終的に一方の鏡像体を収率100%で得ることができる。今まさに社会が必要としている反応である。
2.デラセミ化反応とは?
生体触媒を用いてラセミ体から光学活性体を調製する際の一般的な方法は速度論的光学分割であろう (図1)1)。本法は両鏡像体に対する酵素反応の速度差を利用したものであり、一方の鏡像体が出発物質とは違った構造に変換される。構造が異なれば出発物質とは物理的・化学的性質が異なるため分離が可能で、結局2つの鏡像体を分けたことになる。このような手法を用いれば光学的に純粋な化合物が得られるが、収率は最大でも50%にとどまってしまう。また、反応系には2つの成分が混在することとなるため、必ず分離操作が必要となる。この分離段階が最も煩雑な段階であるといっても過言ではない。
これに対してデラセミ化反応は、酵素がラセミ体基質のうち一方の鏡像体の立体配置のみを認識し、これを立体反転して他方の鏡像体へと変換する。つまり基質と生成物の間で違っているのは立体配置のみということになる。したがって光学的に純粋な化合物を100%の収率にて回収できる。また反応の前後で構造が同じであることから分離操作も必要なく、原子効率も100%というまさに夢のような反応である。デラセミ化反応はラセミ体からの光学活性体調製法としてこれまでの限界を乗り越える究極の方法であるといえる2)。
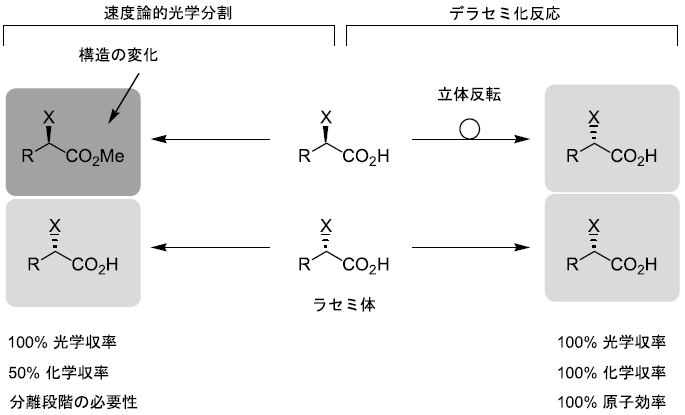
図1 速度論的光学分割とデラセミ化反応の概念図
3.α-メチルカルボン酸のデラセミ化反応
α-メチルカルボン酸に対するデラセミ化反応としてはこれまでいくつかの報告例がある。R体あるいはラセミ体をS体の2-アリールプロパン酸へと変換する生体触媒としてCordyceps militaris3)やラット肝臓抽出物4)が知られている。S体は非ステロイド系抗炎症剤として用いられる。特にラットの場合には反応機構も推定されている (図2)。反応は3段階からなっている。即ち、長鎖脂肪酸に対して作用するアシル-CoAシンセターゼによるチオエステル化、エピメリ化およびチオエステル部分の加水分解である。これらの中でチオエステル化の段階が立体選択的であるため、反応の進行と共に系中の鏡像体比は徐々に一方へと偏っていく。
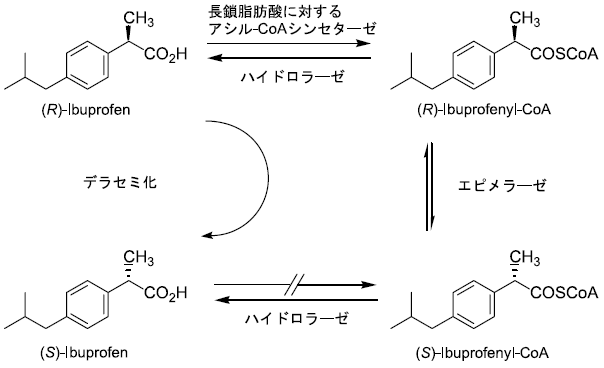
図2 ラット肝臓抽出物を用いたデラセミ化反応推定反応機構
我々は、放線菌の一種Nocardia diaphanozonaria JCM 3208株が2-フェニルプロパン酸 (2-PPA) をデラセミ化し、R体へと変換する能力を有していることを確認した (図3)。この選択性はこれまでの例とは逆であり、R体の新たな調製法として利用できると考えた。この場合、菌体を培養しラセミ体基質を添加するだけというシンプルな操作にて光学活性体が得られた。しかし、本反応は平衡反応でありどちらの鏡像体から出発しても最終的に70%ee 程度に落ち着くことが分かった。また基質特異性も狭く、芳香族環上にフッ素原子が導入されただけでも反応は進行しなくなった。2位の置換基についてもメチル基よりも大きな置換基は受けつけずフッ素原子の導入された2-フルオロフェニル酢酸でのみ反応は進行した。一方、芳香族環と不斉炭素の間に酸素原子を挿入した2-フェノキシプロパン酸 (2-POPA) では高い選択性が発揮された。興味深いことに、本化合物に対する立体選択性は2-PPAに対するそれとは逆転していた。また芳香族環上のp-位はある程度の嵩高さまで許容された5,6)。
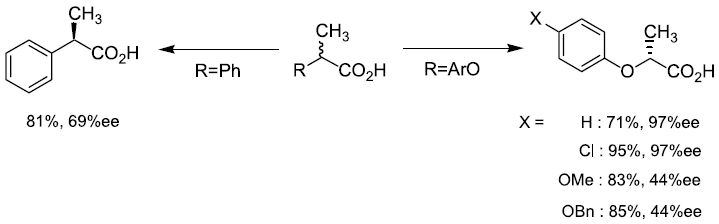
図3 N. diaphanozonariaを用いたα-メチルカルボン酸のデラセミ化反応
しかし、全菌体を用いているため、いくつかの化合物では代謝反応が進行してしまった。そこで、副反応を抑制しデラセミ化反応を優先させる条件の検討を行った7)。例えば2-メチル-3-フェニルプロパン酸は完全に代謝され安息香酸に変換された。その生成経路として脂肪酸のβ-酸化を考えた (図4)。その理由としては全菌体での反応の際、中鎖脂肪酸や安息香酸を添加するとデラセミ化反応が阻害されたこと、また破砕上清を用いて反応を進行させるためには補因子としてATP, CoASH, Mg2+が必要であり、チオエステル体を経由していると予想されたことである。2-メチルカルボン酸のβ-酸化ではメチル基が結合している炭素の立体配置がS の場合のみ代謝をうけるため、エピメリ化酵素が作用することが知られている8)。つまりR体のα-メチンプロトンは引き抜かれ、逆側からプロトンを受け取ることによって立体が反転する。ここで、2-PPAのα-メチンプロトンを重水素化した化合物に対してデラセミ化反応を行うと、その重水素化率は反応の進行と共に低下していくことが分かった。つまりデラセミ化の過程においてもエピメリ化の段階が存在したのである。このことからデラセミ化とβ-酸化は一部共通の反応を利用していると仮定し、それぞれの経路への分かれ道はα, β-不飽和化の段階であると考えた。つまり、エピメリ化酵素の働きによって立体反転を受けた後、速やかにチオエステルが加水分解されればデラセミ化が優先することとなるが、加水分解よりもα, β-不飽和化の方が早いとβ-酸化が進行することとなる。よってα, β-不飽和化酵素を阻害すればデラセミ化が優先すると考えた。そこでα, β-不飽和化を触媒する酵素、アシル-CoAデヒドロゲナーゼの阻害剤である2-ブロモヘキサン酸9)を反応系中に添加し、全菌体にて2-メチル-3-フェニルプロパン酸を作用させた。種々検討の結果、添加濃度を1.2 mMとした際にデラセミ化反応を優先させることができた (71%, 88%ee)。このことから、デラセミ化反応は脂肪酸の代謝経路のひとつであるβ-酸化経路の一部を利用していることが明らかとなった。
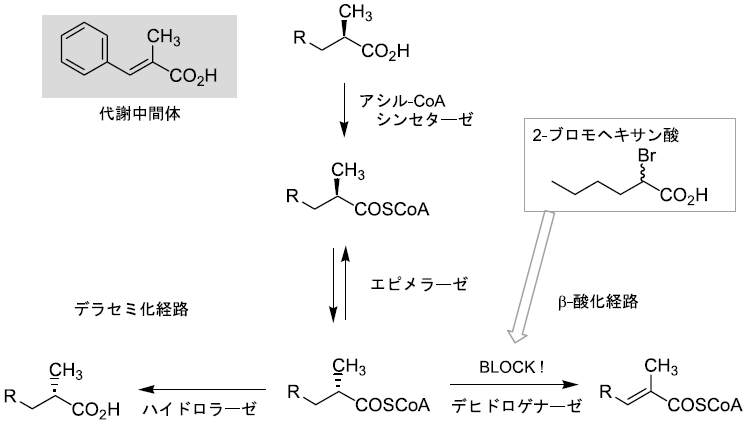
図4 β-酸化とデラセミ化の分かれ道
ところで菌体を破砕すると反応活性は速やかに消失した。つまり反応に関与する酵素は精製に耐えられるほどの安定性を有していなかった。そこで、より安定なデラセミ化酵素系を有する菌株を探索することとした。基質を2- (4-クロロフェノキシ) プロパン酸 (2-CPOPA) としてスクリーニングを行ったところ、新たな微生物を3種見出した。幸いなことにそれらの微生物のうち一株は、菌体を破砕してもデラセミ化活性を示し、また酵素も安定に存在することが分かった。本菌株の有する酵素活性はN . diaphanozonaria と比較すると低いが、2-PPAと2-POPA誘導体に対する立体選択性が同様であったこと、中鎖脂肪酸を添加すると反応が阻害されたことなどから、2種の菌株の酵素系は類似していると予想している (図5)。破砕上清を用いた場合には補因子としてATP, CoASH, Mg2+を要求したためチオエステル化を指標に酵素の精製およびクローニングを試みた。得られた酵素はバクテリア由来のアシル-CoAシンセターゼと非常に高い相同性を示すことがわかった。精製酵素を利用して2-CPOPAとの反応を試みたところ、反応はS体選択的に進行した (図6)。本菌株でも図2と同様にチオエステル体を中間体とする複合反応機構でデラセミ化が実現されると考えている。
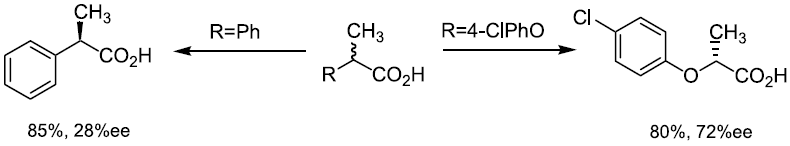
図5 スクリーニング菌株を用いたα-メチルカルボン酸のデラセミ化反応
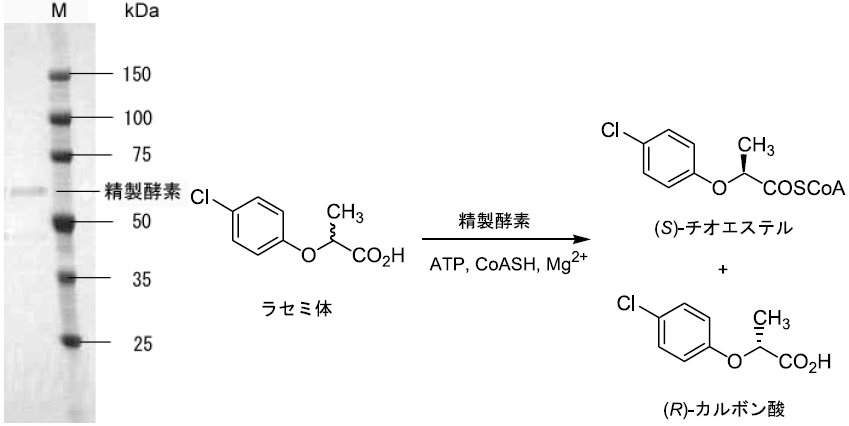
図6 チオエステル化酵素の精製と反応性
4.α-アミノ酸のデラセミ化反応
一般に微生物はD-アミノ酸をそのままエネルギー源として利用することはできない。そのためD-フェニルアラニンを唯一の炭素源とする無機塩培地で生育する微生物はアミノ酸の立体配置をL体へと変換できるのではないかと考えた。立体反転過程の有無を色の変化で認識できるようなスクリーニング系を組み立てることで効率よく目的の活性を有する菌体を得ることができた10)。例えばN. diaphanozonaria や窒素固定菌の一種であるSinorhizobium meliloti ATCC 51124株、Mesorhizobium loti MAFF 303099株などである。特にN. diaphanozonaria はフェニルアラニン誘導体だけでなくフェニルグリシンや2-アミノヘプタン酸、tert-ロイシンなど幅広い基質に対して活性を示した (図7)。
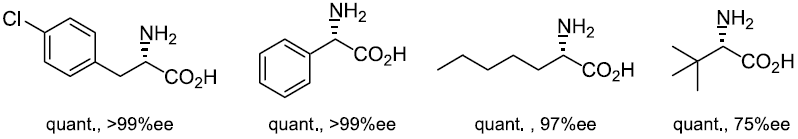
図7 N. diaphanozonariaを用いたα-アミノ酸のデラセミ化反応
S. melilotiに対して4-クロロフェニルアラニンを作用させた際の経時変化を追跡した (図8)。反応中、α-ケト酸とアミノ酸の濃度の和はほとんど変化がないにもかかわらず、D体濃度は低下し、それに代わってL体濃度が上昇することが分かる。またα-ケト酸濃度の一時的な上昇も確認されたことから、中間体としてα-ケト酸を経由していると考えた。実際、α-ケト酸を基質とした場合には、反応の進行にしたがって濃度は低下し、それに伴ってL体のアミノ酸濃度が上昇することを確認している。
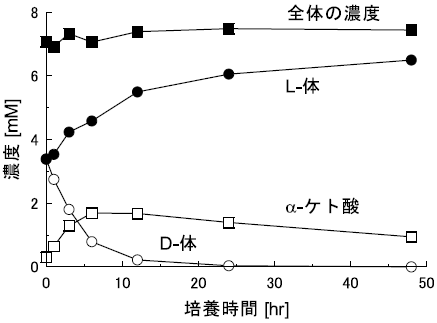
図8 S. melilotiを用いた4-クロロフェニルアラニンのデラセミ化反応
次に破砕上清を用いた検討を行った。菌体を破砕しても高いデラセミ化活性が残存していたが、透析操作を行うとその活性は著しく低下したことから反応には補因子が必要であることがわかった。検討の結果、アミノ酸からα-ケト酸への反応はD体選択的であり、補因子として電子受容体であるフェナジンメタサルフェート (PMS) を要求することが分かった。次に、α-ケト酸からアミノ酸への変換反応を確認したところピリドキサールリン酸 (PLP) とL-グルタミン酸を添加した際に反応が進行しL体のアミノ酸が得られた。最終的に、これら2種類の補因子を添加すれば破砕上清を透析しても各種アミノ酸に対して効率よくデラセミ化反応を進行させることができるようになった。このように、アミノ酸のデラセミ化反応はα-ケト酸を中間体とし、D体選択的脱アミノ化反応とL体選択的アミノ基転位反応という2段階にて実現されていることを明らかにした (図9)。
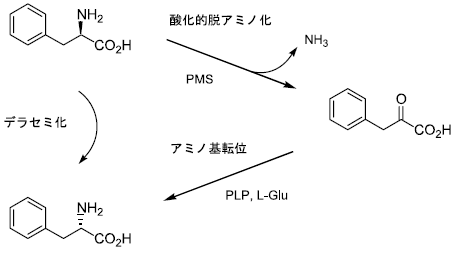
図9 α-アミノ酸に対するデラセミ化反応機構
さて、S. melilotiについては既に全塩基配列が明らかになっておりORFもデータベース上にて公開されている。これらの情報を基に分岐鎖アミノ酸に対するアミノ基転位酵素 (BCAAT) を大菌にて発現させ、全菌体にてデラセミ化反応を進行させることを試みた11)。本組換え体を用いて反応を実現するためにはアミノ基酸化酵素が別に必要であるが、これは宿主が有しているD-アミノ酸デヒドロゲナーゼ (DadA) を利用することとした。DadAはLB培地で培養を行った場合には弱い活性しか示さないが、L-アラニンを含む無機塩培地中で培養を行うと酵素の発現が誘導される。そこで、L-アラニンを含む無機塩培地中で培養を行うことによってDadAを、IPTGを添加することによってBCAATを誘導し、デラセミ化に必要な2種類の酵素を同時に活性化することとした。本菌を用い4-クロロフェニルアラニンを作用させたところ確かにデラセミ化反応が効率よく進行することが分かった (図10)。BCAATを誘導しなかった場合には脱アミノ化反応は進行するものの、α-ケト酸を生成した段階で反応は止まり、デラセミ化はほとんど進行していない (図11)。このように1種類の外来遺伝子にて形質転換された大腸菌にも関わらずデラセミ化という2つの酵素を必要とする新規な活性を大腸菌に付与することに成功した。これは大腸菌をタンパク質合成のための工場としてみるだけでなく、大腸菌の有している機能を目的の反応を実現するためのプロセスに組み込んで利用するという考え方によるものであり、今後の生体触媒反応に新たな方向性を示すものといえる。
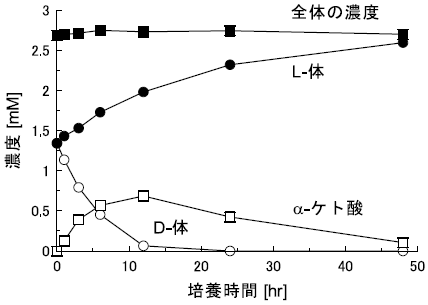
図10 組換え大腸菌を用いた4-クロロフェニルアラニンのデラセミ化反応: BCAAT誘導時
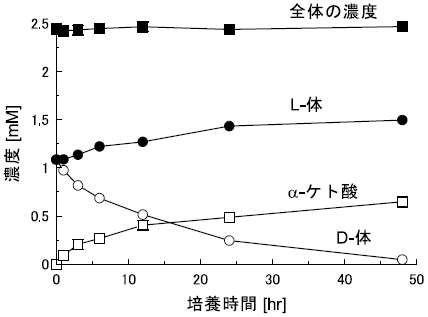
図11 組換え大腸菌を用いた4-クロロフェニルアラニンのデラセミ化反応: BCAAT非誘導時
5.まとめ
ラセミ体から光学活性体を調製する方法として、デラセミ化反応を紹介した。α-メチルカルボン酸に関する検討では全菌体のN . diaphanozonariaを用いてR体の2-フェノキシプロパン酸誘導体を高収率かつ高い光学収率で調製する方法を確立した。機構解析では反応が脂肪酸の代謝経路の一部を利用して実現されていることを明らかにした。またデラセミ化に関連する酵素のうち立体選択的なチオエステル化を触媒する酵素の精製にも成功した。α-アミノ酸に対する検討ではS. melilotiを用いて反応機構解析を行い、α-ケト酸を中間体とする2段階反応にて実現されていることを明らかにできた。さらにL体選択的なアミノ基転位酵素 (BCAAT) を大量発現させた組換え大腸菌を用い、また培養条件を工夫することによって効率よくデラセミ化反応を実現する系を構築することに成功した。
謝辞
ここで紹介したデラセミ化反応は、私が慶應義塾大学理工学部生命情報学科、酵素有機化学研究室在学中に検討した内容であり、研究を行うに当たりましては、太田博道教授より多大なご指導を賜りました。ここに深く感謝いたします。
文献
1) Ghanem, A., Aboul-Enein, H. Y.: Chirality, 17, 1 (2005).
2) Turner, N. J.: Curr. Opin. Chem. Biol., 8, 114 (2004).
3) Rhys-Williams, W., Thomason, M. J., Hung, Y.- F., Hanolon, G. W., Lloyd, A. W.: Chirality, 10, 528 (1998).
4) Shieh, W.-R., Chen, C.-S.: J. Biol. Chem., 268, 3487 (1993).
5) Kato, D.-i., Mitsuda, S., Ohta, H.: Org. Lett., 4, 371 (2002).
6) Kato, D.-i., Mitsuda, S., Ohta, H.: J. Org. Chem., 68, 7234 (2003).
7) Kato, D.-i., Miyamoto, K., Ohta, H.: Tetrahedron: Asymmetry, 15, 2965 (2004).
8) Ferdinandusse, S., Denis, S., Ijlst, L., Dacremont, G., Waterham, H. R.: J. Lipid Res., 41, 1890 (2000).
9) Haeffner-Gormley, l., Cummings, J. G., Thorpe, C.: Arch. Biochem. Biophys., 317, 479 (1995).
10) Kato, D.-i., Miyamoto, K., Ohta, H.: J. Mol. Cat.B: Enzym., 32, 157 (2005).
11) Kato, D.-i., Miyamoto, K., Ohta, H.: Biocat. Biotransform., in press.
![]()